The post Innovationsfonds: Förderung für digitale Gesundheitslösungen appeared first on QuickBird Medical.
]]>Der Markt für Selbstzahler ist im deutschen Gesundheitswesen überschaubar, und der Weg in die Erstattung durch Krankenkassen herausfordernd.
Zwar existieren etablierte Erstattungswege wie die Aufnahme ins DiGA-Verzeichnis oder eine individuelle Vergütung über Selektivverträge, doch diese Zugangswege können langwierig, komplex und kostenintensiv sein.
Daher lohnt es sich, auch alternative Pfade zur Finanzierung zu betrachten, während man auf den Sprung in die Regelversorgung hinarbeitet. Eine solche Alternative für die Finanzierung und Erstattung digitaler Gesundheitslösungen ist der Innovationsfonds nach § 92a SGB V.
In diesem Fachartikel erklären wir …
- welche Möglichkeiten der Innovationsfonds bietet,
- welche Voraussetzungen für eine Förderung erfüllt sein müssen,
- wie hoch die Erfolgschancen bei Software-Produkten sind,
- und für welche Arten von digitalen Konzepten eine Antragstellung möglich ist.
Update – Dezember 2025: Im Dezember 2025 zog die „BASYS“ eine Bilanz über zehn Jahre „Neue Versorgungsformen“ im Innovationsfonds. Die wichtigsten Ergebnisse fassen wir in Kapitel 6 zusammen.
Inhaltsverzeichnis
- 1. Definition des Innovationsfonds
- 2. Unterschied zwischen Förderung & Erstattung
- 3. Voraussetzungen für die Förderung durch den Innovationsfonds
- 4. Das Antragsverfahren des Innovationsfonds
- 5. Für welche Software-Produkte kommt eine Innovationsfonds-Förderung infrage
- 6. Zwischenbilanz nach 10 Jahren Innovationsfonds: Was kommt wirklich in der Versorgung an?
- 7. Fazit: Lohnt sich der Innovationsfonds für Hersteller medizinischer Software?
- 8. Wo finde ich weitere Informationen zum Innovationsfonds?
1. Definition des Innovationsfonds
1.1 Was ist der Innovationsfonds des G-BA?
Der Innovationsfonds wurde Anfang 2016 durch das GKV-Versorgungsstärkungsgesetz ins Leben gerufen und ist beim Gemeinsamen Bundesausschuss (G-BA) angesiedelt.
Er dient dem Ziel, neue Versorgungsformen und Versorgungsforschung zu fördern, die über die bisherige Regelversorgung hinausgehen. Konkret sollen innovative Ansätze im Gesundheitswesen wissenschaftlich erprobt werden, um die Versorgung effizienter, patientenzentrierter und qualitativ besser zu gestalten. Der Fonds hat sich seither als zentrales Instrument etabliert, um (digitale) Innovationen im Gesundheitswesen unter realen Bedingungen zu testen.
1.2 Wie hoch ist das verfügbare Fördervolumen?
Seit dem Start stehen jährlich rund 200 Millionen Euro an Fördermitteln zur Verfügung. Ursprünglich war der Fonds befristet (bis 2019, später verlängert bis 2024), wurde jedoch nach positiver Evaluation entfristet und ab 2025 verstetigt.
Der Großteil des Budgets (160 Mio. €) fließt in Projekte zu neuen Versorgungsformen, während 40 Mio. € für Versorgungsforschung reserviert sind.
1.3 Wer verwaltet den Innovationsfonds?
Zuständig ist der Innovationsausschuss beim Gemeinsamen Bundesausschuss (G-BA), der in regelmäßigen Förderbekanntmachungen, thematische Schwerpunkte und Kriterien festlegt und über die Anträge entscheidet.
Projekte können typischerweise eine Laufzeit von bis zu drei (manchmal auch vier) Jahren haben und werden wissenschaftlich begleitet.
Nach Abschluss eines Projekts werden die Ergebnisse ausgewertet und an relevante Gremien weitergeleitet, um zu prüfen, ob die Innovation in die Regelversorgung übernommen werden sollte.
Der Innovationsausschuss setzt sich aus Vertretern der Trägerorganisationen des G-BA (Krankenkassen, Kassenärztliche Vereinigungen, Krankenhäuser) sowie dem Bundesministerium für Gesundheit und dem Bundesministerium für Forschung zusammen. Er wird zudem von unabhängigen Experten aus Wissenschaft und Versorgung.

Zusammensetzung des Innovationsausschusses
1.4 Wer ist beteiligt?
Typischerweise schließen sich für Innovationsfondsprojekte verschiedene Akteure im Gesundheitswesen zu Konsortien zusammen. Dazu gehören z. B.:
- Krankenkassen
- Krankenhäuser und Universitätskliniken
- ärztliche Netzwerke oder MVZ
- Forschungseinrichtungen
- Private Unternehmen (z.B. im Bereich Digital Health)
Gemeinsam entwickeln sie ein Versorgungskonzept, das dann für mehrere Jahre unter Alltagsbedingungen erprobt wird. Digitale Gesundheitslösungen sind dabei oft ein zentraler Bestandteil – etwa zur Koordination, Kommunikation oder zur Diagnostik.
1.5 Förderkategorien im Überblick
Der Innovationsfonds unterscheidet zwei Förderbereiche gemäß § 92a SGB V:
- Kategorie 1: Neue Versorgungsformen
- Kategorie 2: Versorgungsforschung
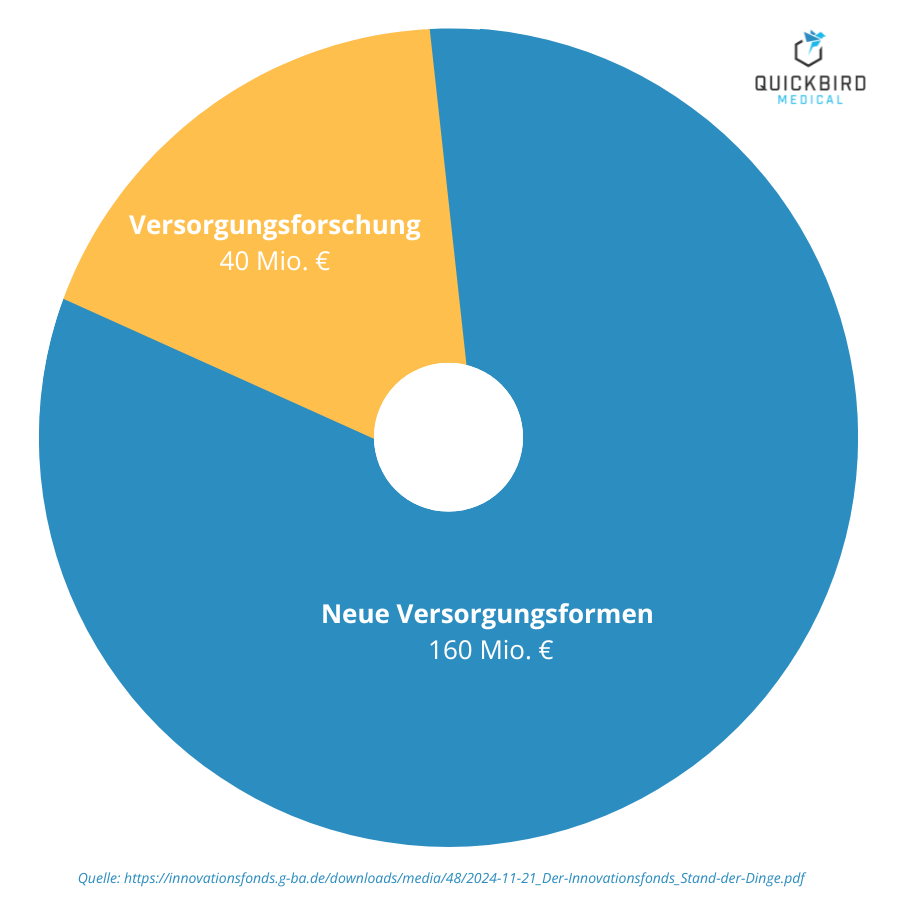
Förderkategorien und Fördervolumen des Innovationsfonds (2025)
1.5.1 Kategorie 1: Neue Versorgungsformen
Hierunter fallen Projekte, die neue Versorgungsmodelle oder organisatorische Abläufe in der Patientenversorgung erproben. Digitale Gesundheitsanwendungen sind dabei oft Bestandteil des Konzepts – etwa Telemedizin-Plattformen, digitale Kommunikations-Tools oder KI-gestützte Diagnose- und Therapiesteuerung.
Wichtig: Diese Projekte finden unter Alltagsbedingungen in der Versorgung statt – etwa als Pilotprogramme mit echten Leistungserbringern und Patienten. Die digitale Lösung wird dabei immer in ein Versorgungskonzept eingebettet, etwa um Prozesse zu optimieren oder Sektorengrenzen zu überwinden.
Begleitet wird das Ganze durch eine wissenschaftliche Evaluation, um die Effekte auf Versorgung und Patientenversorgung messbar zu machen.
Für Hersteller medizinischer Software ist diese Kategorie besonders relevant, da hier speziell komplett neue Lösungen und Strukturen evaluiert werden.
1.5.2 Kategorie 2: Versorgungsforschung
In diesem Bereich geht es nicht um die Umsetzung neuer Versorgungsmodelle, sondern um die Analyse der bestehenden Versorgungsrealität. Gefördert werden z. B. wissenschaftliche Studien zur Evaluation von G-BA-Richtlinien, Leitlinienentwicklungen oder gesundheitsökonomische Analysen.
Für Hersteller ist diese Kategorie nur dann relevant, wenn die eigene digitale Lösung bereits Teil der Regelversorgung ist und im Rahmen einer Studie evaluiert werden soll.
Wenn dagegen das Produkt erst noch in die Versorgung gebracht werden soll, fällt das Vorhaben nicht in diese Kategorie, sondern gehört zu den „Neuen Versorgungsformen“.
Hinweis: In diesem Artikel konzentrieren wir uns ausschließlich auf den Förderbereich „Neue Versorgungsformen“, da dieser für Hersteller medizinischer Software in der Praxis deutlich relevanter ist als die ebenfalls mögliche Förderung im Bereich Versorgungsforschung.
2. Unterschied zwischen Förderung & Erstattung
Wenn wir über die Finanzierung digitaler Gesundheitslösungen sprechen, ist es wichtig, den Unterschied zwischen Förderung und Erstattung zu verstehen. Eine Erstattung – wie etwa bei DiGA – bedeutet: Die Anwendung ist im Leistungskatalog der GKV enthalten, und jede Nutzung wird von der Krankenkasse bezahlt. Das schafft planbare Umsätze und ist Teil der Regelversorgung.
Der Innovationsfonds funktioniert anders: Hier handelt es sich nicht um eine dauerhafte Leistung, sondern um eine zeitlich begrenzte Projektförderung.
Für Hersteller von Software-Produkten bedeutet eine Förderung über den Innovationsfonds konkret: Die Kosten für die Software-Implementierung im Projekt sind gedeckt – sei es als Auftragnehmer oder Konsortialpartner. Und auch wenn die Software noch nicht offiziell von den Kassen erstattet wird, fließt bereits Geld – über den G-BA-Fördertopf.
Gleichzeitig entstehen dabei echte Versorgungsdaten, die später als Grundlage für weitere Erstattungswege dienen können: etwa Selektivverträge oder eine Vergütung über das DRG-System.
3. Voraussetzungen für die Förderung durch den Innovationsfonds
3.1 Welche Projekte werden gefördert?
Gefördert werden neue Versorgungsformen, die
- insbesondere die Weiterentwicklung der sektorenübergreifenden Versorgung zum Ziel haben:
- Überwindung der Trennung der Sektoren
- Optimierung intersektoraler Schnittstellen
- Weiterentwicklung der selektivvertraglichen Versorgung
- ein tragfähiges Evaluationskonzept vorweisen und
- ein hinreichendes Potenzial für eine dauerhafte Aufnahme in die Versorgung (Umsetzungspotenzial) aufweisen.
Weitere Informationen dazu finden Sie hier.
3.2 Welche Projekte werden nicht gefördert?
Nicht jedes Projekt ist für eine Förderung durch den Innovationsfonds geeignet. Es gibt eine Reihe von Ausschlusskriterien, die Antragsteller kennen sollten, bevor sie Zeit und Ressourcen investieren. Hier die wichtigsten Punkte:
- Wirtschaftlich getriebene Produktentwicklung: Der Innovationsfonds ist kein Förderprogramm für klassische Produktentwicklung mit direktem kommerziellem Ziel. Projekte, bei denen Unternehmen der gewerblichen Wirtschaft ein unmittelbares wirtschaftliches Interesse am Ergebnis haben, sind ausgeschlossen – etwa die Entwicklung oder Erprobung eines Produkts mit dem Ziel, es anschließend direkt zu verkaufen.
- Klassische F&E, Zulassungs- oder Arzneimittelstudien
- Forschung und Entwicklung von Medizinprodukten oder Arzneimitteln
- Klinische Prüfungen nach EU-Medizinprodukteverordnung (EU) 2017/745
- Leistungsbewertungsprüfungen für In-vitro-Diagnostika
- AMNOG-Studien zur frühen Nutzenbewertung von Arzneimitteln
- Studien zur Erprobung neuer Untersuchungs- und Behandlungsmethoden (§ 137e SGB V)
- DiGA- und DiPA-Nachweisstudien: Auch Studien zum Nachweis positiver Versorgungseffekte im Rahmen von DiGA (§ 33a SGB V) oder DiPA (§ 40a SGB XI) sind nicht förderfähig. Diese Nachweise müssen separat über das BfArM bzw. entsprechende Verfahren erbracht werden und fallen nicht in den Zuständigkeitsbereich des Innovationsfonds.
- Projekte in der Umsetzung oder mit anderer Förderung: Bereits gestartete Projekte oder Vorhaben, die zum Zeitpunkt der Antragstellung bereits anderweitig (z. B. aus öffentlichen Mitteln) gefördert werden, sind ausgeschlossen. Der Innovationsfonds fördert keine Doppelstrukturen oder rückwirkenden Maßnahmen.
- Doppelte Bewerbung ausgeschlossen: Für jede der in einem Jahr ausgeschriebenen Förderbekanntmachungen zu „Neuen Versorgungsformen“ darf man sich nur einmal Eine doppelte oder parallele Antragstellung mit gleichem Projektansatz ist nicht zulässig.
4. Das Antragsverfahren des Innovationsfonds
4.1 Welche Antragsverfahren gibt es?
Der Innovationsfonds unterscheidet im Bereich der Neuen Versorgungsformen vier verschiedene Förderformate. Diese unterscheiden sich im Hinblick auf Themenvorgabe, Ablaufstruktur (ein- vs. zweistufig) und Projektdauer:
- Themenoffene Förderung – Einstufig lang
In diesem Verfahren wird ein vollständiger Antrag direkt eingereicht. Das Projekt muss dabei bereits umfassend geplant sein, inklusive Evaluationskonzept, Zeit- und Finanzplan sowie Konsortium.
- Themenoffene Förderung – Einstufig kurz
Dieses Format richtet sich an kleinere, schneller startende Projekte mit einer Laufzeit von maximal 24 Monaten. Auch hier erfolgt die Antragstellung direkt und vollständig in einem Schritt.
- Themenspezifische Förderung – Zweistufig lang
Bei diesem Verfahren gibt es zwei Phasen:
- Stufe 1: Konzeptphase – Einreichung einer Ideenskizze zu einem vorgegebenen Thema.
- Stufe 2: Vollantrag – Eingereicht wird ein kompletter Förderantrag mit vollständig ausgearbeitetem Versorgungskonzept, konkreten Versorgungszielen und Umsetzungsplanung, validiertem Evaluationskonzept, Zeit- und Finanzplan und verbindlichen Zusagen aller Konsortialpartner.
Dieses Verfahren eignet sich für komplexe Projekte mit größerem Planungsbedarf.
- Themenoffene Förderung – Zweistufig lang
Auch hier ist das Verfahren zweistufig:
- Stufe 1: Konzeptphase – Ideenskizze ohne thematische Vorgabe.
- Stufe 2: Vollantrag – wie bei der themenspezifischen Variante.
Damit bietet diese Förderlinie Freiraum für innovative Ideen mit größerem Entwicklungsaufwand.
Hinweis: Alle Verfahren setzen voraus, dass die Antragsunterlagen vollständig und fristgerecht eingereicht werden. Welche Unterlagen erforderlich sind (z. B. Projektbeschreibung, Konsortialvertrag, Finanzierungsplan, Ethikkonzept), ist detailliert in den jeweiligen Förderbekanntmachungen geregelt.
→ Link zu den aktuellen Ausschreibungen:
https://innovationsfonds.g-ba.de/foerderbekanntmachungen/
4.2 Welches Antragsverfahren passt zu meinem Projekt?
Wann welcher Antrag am ehesten in Frage kommt, hängt vom Reifegrad der Projektidee ab.
- Reifer Projektstand:
- Projektidee ist vollständig ausgearbeitet.
- Es liegt ein ausgereiftes Versorgungskonzept vor.
- Konsortialpartner (z. B. Kassen, Versorgungseinrichtungen) sind verbindlich eingebunden.
- Evaluationskonzept, Zeitplan und Budget sind konkret geplant.
➔ einstufiges Verfahren
- Weniger ausgereifter Projektstand:
- Grundidee ist vorhanden, aber noch nicht detailliert ausgearbeitet.
- Es fehlt noch an validierten Partnern, konkreter Evaluation oder präziser Umsetzungsplanung.
- Ziel ist es, in einer 6-monatigen Konzeptphase (Stufe 1) die Projektidee zu schärfen.
- Danach kann ein Vollantrag in Stufe 2 gestellt werden.
➔ zweistufiges Verfahren
Vergleich des einstufigen und zweistufigen Verfahrens:
| Kriterium | Einstufig (lang/kurz) | Zweistufig (lang) |
| Projektidee |
vollständig ausgereift | noch in Konzeptionsphase |
| Partner | konkrete Zusagen | noch unklar / in Vorbereitung |
| Zeitbedarf vor Start | kürzer, kein Zwischenschrit | 6-monatige Konzeptphase erforderlich |
| Förderung Konzeptphase | nein | ja, Stufe 1 wird gefördert |
| Bürokratischer Aufwand | geringer | höher (2 Antragsrunden) |
| Flexibilität während Planung | geringer | höher (Konzept kann noch angepasst werden) |
4.3 Was sind die Anforderungen für einen Antrag?
Damit ein Projekt überhaupt förderfähig ist, müssen einige grundlegende Rahmenbedingungen erfüllt sein. Diese gelten unabhängig vom Antragsverfahren:
- Rechtskonforme Umsetzung: Die geplante Versorgungsform muss sich im Rahmen der bestehenden gesetzlichen Grundlagen bewegen – das heißt: Sie darf nichts umsetzen, was z. B. ärztlichen Berufsordnungen oder dem SGB V widerspricht.
- Datenschutz & Ethik: Die Verarbeitung von Gesundheitsdaten muss den Anforderungen der DSGVO entsprechen. Ebenso wird erwartet, dass ethische Standards eingehalten werden – etwa bei Studien mit Patienten oder bei sensiblen Datenverarbeitungen.
- Wissenschaftlicher Anspruch: Das Projekt muss wissenschaftlich fundiert aufgebaut sein – sowohl im Versorgungsteil als auch bei der Evaluation. Dabei gelten Standards aus der Versorgungsforschung und empirischen Studienplanung.
- Interoperabilität & Schnittstellen: Digitale Lösungen müssen mit anderen Systemen im Gesundheitswesen kompatibel sein. Besonders relevant ist die Anbindung an die Telematikinfrastruktur (TI) der gematik.
- Ergebnisse zugänglich machen: Die Evaluationsergebnisse müssen transparent veröffentlicht werden – unabhängig davon, ob das Projekt erfolgreich war oder nicht. Es geht nicht darum, interne Pläne oder Skizzen offenzulegen, sondern um eine nachvollziehbare Veröffentlichung der Ergebnisse nach Projektende.
- Beteiligung an Meta-Evaluationen: Die Ergebnisse einzelner Projekte sollen in eine Gesamtbewertung der Förderstrategie einfließen. Dazu kann der G-BA auffordern, an übergreifenden Evaluationsformaten mitzuwirken – etwa für die Analyse, welche Arten von Innovationen sich langfristig bewähren.
4.4 Was sind die Kriterien zur Förderung?
Sofern alle eben genannten, grundlegenden Fördervoraussetzungen erfüllt sind, erfolgt die fachliche Bewertung anhand der Förderkriterien:
- Relevanz
- Verbesserung der Versorgung
- Verbesserung der Versorgungsqualität und/oder Versorgungseffizienz
- Behebung von Versorgungsdefiziten
- Optimierung der Zusammenarbeit innerhalb und zwischen verschiedenen Versorgungsbereichen, Versorgungseinrichtungen und Berufsgruppen
- Interdisziplinäre und fachübergreifende Versorgungsmodelle
- Umsetzungspotenzial
- Übertragbarkeit der Erkenntnisse, insbesondere auf andere Regionen oder Indikationen
- Evaluierbarkeit: methodische und wissenschaftliche Qualität des Evaluationskonzepts
- Machbarkeit des Projekts in der Laufzeit
- Verhältnismäßigkeit von Implementierungskosten und Nutzen
- Patientenbeteiligung
Die Förderkriterien werden jedes Jahr in den jeweiligen Förderbekanntmachungen detailliert beschrieben.
Die Entscheidung selbst wird zweistufig getroffen:
- Evaluation durch Experten-Pool: Zuerst bewerten unabhängige Expertinnen und Experten aus dem sogenannten „Expertenpool“ die eingereichten Anträge. Der Expertenpool wird alle zwei Jahre neu gewählt und bestehend aus “Personen, die praktische oder wissenschaftliche Erfahrung aus der (digitalen) Gesundheitsversorgung mitbringen und über Sektoren- wie Berufsgrenzen” verfügen.
- Entscheidung durch G-BA: Auf Basis des Antrags und der Empfehlungen des Expertenpools entscheidet der Innovationsausschuss des G-BA, welche Projekte eine Förderung erhalten.
4.5 Beispiel für ein Innovationsfonds-gefördertes Projekt: INTEGRATE-ATMP
Ein konkretes Beispiel für die erfolgreiche Förderung einer digitalen Gesundheitslösung ist das Projekt INTEGRATE-ATMP, das durch das Universitätsklinikum Heidelberg initiiert wurde. QuickBird Medical hat in diesem Projekt die technische Umsetzung der Software-Plattform in enger Zusammenarbeit mit allen involvierten Akteuren übernommen und erfolgreich abgeschlossen.
Dank einer Förderung durch den Innovationsfonds des Gemeinsamen Bundesausschusses (G-BA) von 13,6 Millionen Euro ebnet INTEGRATE-ATMP den Weg für harmonisierte Strukturen in der Patientenversorgung. Die telemedizinische Kommunikationsplattform ermöglicht eine strukturierte und direkte Kommunikation zwischen Kliniken, behandelnden ÄrztInnen und den PatientInnen. Ziel ist es, Patienten in ihrem Therapiealltag und deren Behandlungszentren in ihrem Versorgungsalltag zu entlasten. Hierfür wurde eine Telekommunikations-Plattform entwickelt.
Mehr Informationen dazu finden Sie hier: https://quickbirdmedical.com/project/integrate-atmp/
Weitere Beispiele für Innovationsfond-geförderte Projekte:
- BlenCon – Blended Consultation
- Rise-uP – Rückeninnovative Schmerztherapie mit e-Health
- Telenotarzt Bayern
- TELnet@NRW – Telemedizinisches, intersektorales Netzwerk
- WELCOME – Digitale Überleitung in die poststationäre Versorgung
4.6 Was passiert nach Projektende?
Am Ende jedes geförderten Projekts steht für den G-BA eine zentrale Frage im Raum: Hat sich die Idee bewährt – und gehört sie dauerhaft in die Regelversorgung?
Für diese Prüfung stehen dem G-BA drei Monate nach Einreichen des Evaluationsberichts zur Verfügung. Die Entscheidung wird als offizieller Beschluss auf der Website veröffentlicht – inkl. Vorschlag, wie und durch wen eine Umsetzung erfolgen kann.
Typischerweise sind Einrichtungen der Selbstverwaltung zuständig, z. B. der GKV-Spitzenverband oder die Kassenärztliche Bundesvereinigung (KBV). Auch deren Rückmeldungen sind öffentlich einsehbar – was nachvollziehbar macht, ob ein Projekt wirklich Wirkung entfaltet hat.
Eine zentrale Liste, in der man auf einen Blick sehen kann, welche Projekte in die Regelversorgung übernommen wurden oder eine Umsetzungsempfehlung erhalten haben, gibt es derzeit leider nicht. Seit 2024 sind die angesprochenen Stellen aber verpflichtet, dem G-BA mitzuteilen, was aus den Projektvorschlägen geworden ist. Diese Rückmeldungen sind auf den einzelnen Projektseiten zu finden – zum Beispiel beim Projekt Sisyphos.
Die Überführung in die Versorgung kann auf verschiedenen Wegen passieren: über gesetzliche Änderungen (z. B. im SGB V), über Verträge, über neue Richtlinien oder durch Empfehlungen.
Bisher wurde nur bei einem einzigen Projekt ein direkter Beschluss zur Aufnahme in die Versorgung getroffen – dieser musste jedoch nachträglich zurückgezogen werden, weil das Projektteam seine Ergebnisse korrigiert hat. Die Pressemitteilungen dazu findet man hier.
5. Für welche Software-Produkte kommt eine Innovationsfonds-Förderung infrage
Nicht jede digitale Gesundheitslösung ist gleichermaßen für den Innovationsfonds geeignet. Angesichts des beschriebenen Aufwands und der Voraussetzungen lohnt sich eine Bewerbung vor allem für bestimmte Produktarten und Entwicklungsstadien. Die folgenden Unterkapitel zeigen dies beispielhaft auf.
5.1 Komplexe Versorgungslösungen – keine einzelnen Software-Produkte
Produkte, die tief in Versorgungsprozesse eingreifen, stehen stärker im Fokus als einzelne entkoppelte Software-Produkte. Beispiele: Telemedizinische Netzwerklösungen, sektorübergreifende Plattformen, digitale Systeme zur Koordination zwischen Haus- und Fachärzten oder Klinik und ambulant. Solche Lösungen entfalten ihren Wert erst im Zusammenspiel mehrerer Akteure – ideal für ein Konsortium. Einzelne Apps dagegen (etwa eine reine Selbstmanagement-App ohne Einbindung von Ärzten) haben weniger Anknüpfungspunkte für ein Versorgungsmodell und sind daher seltener Gegenstand von Innovationsfonds-Projekten.
5.2 Produkte mit hohem Evidenzbedarf
Wenn eine Software neuartig ist und von Entscheidungsträgern noch skeptisch beäugt wird, kann ein Innovationsfondsprojekt die nötige Evidenz liefern. Insbesondere bei KI-basierten Diagnosetools, digitalen Therapien für schwerwiegende Erkrankungen oder Software als Medizinprodukt hoher Risikoklasse (IIb/III) gibt es oft hohe Hürden für die Erstattung. Hier kann die wissenschaftliche Begleitung im Rahmen der Förderung den Beleg erbringen, dass das Produkt sicher und effektiv ist. Danach fällt es leichter, z.B. einen positiven G-BA-Beschluss oder einen Eintrag ins Hilfsmittelverzeichnis zu erlangen.
5.3 Nischenindikationen und seltene Krankheiten
Digitale Anwendungen für seltene Erkrankungen oder sehr kleine Patientengruppen sind im DiGA-Fast-Track schwer refinanzierbar, finden aber im Innovationsfonds eine Möglichkeit zur Erprobung. Bei seltenen Erkrankungen stehen oft hohe Entwicklungskosten einem begrenzten Markt gegenüber. Der Innovationsfonds kann hier einspringen und Modellvorhaben finanzieren, die Versorgungslösungen für Orphan Diseases testen. Ein Beispiel ist das Konsortialprojekt TRANSLATE-NAMSE, bei dem mehrere Unikliniken und Kassen zusammenarbeiten, um die Versorgung von Menschen mit seltenen Erkrankungen zu verbessern – gefördert 42 Monate lang mit rund 13,4 Millionen Euro. In solch einem Fall dient der Innovationsfonds quasi als Testlabor für Nischenanwendungen, die sonst aufgrund geringer Fallzahlen keine Chance auf Finanzierung hätten.
5.4 Versorgungsmanagement-Tools
Versorgungsmanagement-Tools – wie Nachsorge-Apps, Triage-Systeme oder Medikationsmanagement-Plattformen – bieten oft einen indirekten Nutzen für das Gesundheitssystem, sind aber im Regelbetrieb schwer umzusetzen. Da solche Lösungen primär Folgeerkrankungen verhindern oder Prozesse effizienter gestalten, passen sie nicht immer in bestehende Vergütungsstrukturen. Ein prominentes Beispiel ist das Medikationsmanagement-Projekt AdAM für Polypharmazie-Patienten: Initiiert von einer Krankenkasse und einer Kassenärztlichen Vereinigung, erhielt es rund 16 Millionen Euro Anschubfinanzierung aus dem Innovationsfonds.
6. Zwischenbilanz nach 10 Jahren Innovationsfonds: Was kommt wirklich in der Versorgung an?
Im Dezember 2025 veröffentlichte die BASYS Beratungsgesellschaft für angewandte Systemforschung eine Auswertung der ersten zehn Jahre „Neue Versorgungsformen“ im Innovationsfonds. Die Ergebnisse geben einen aufschlussreichen Einblick in die bisherige Bilanz und sind auch für Hersteller digitaler Gesundheitslösungen relevant.
Im Förderbereich „Neue Versorgungsformen“ (NVF) wurden in den ersten zehn Jahren 115 von 253 Projekten abgeschlossen. Von diesen 115 Projekten:
- 29 Projekte erhielten eine Prüfbitte zur Überführung in die Regelversorgung
- 30 Projekte wurden mit ihren Erkenntnissen an Fachgesellschaften weitergegeben
- 56 Projekte bekamen keine Empfehlung

Bilanz der abgeschlossenen Projekte mit Neuen Versorgungsformen (NVF) nach 10 Jahren Innovationsfonds-Förderung
Wichtig: „Keine Empfehlung“ heißt nicht automatisch „ohne Nutzen“, die Ergebnisse können trotzdem helfen, Versorgungsmodelle und deren Kosteneffizienz weiterzuentwickeln.
Spannend wird es beim Outcome: Unter den 29 Projekten mit Umsetzungsempfehlung gibt es fünf mit nachgewiesener, signifikanter Kosteneinsparung:
- CoCare mit statistisch signifikant 1,6 eingesparten Krankenhaustagen (sektorübergreifende Koordination von Leistungen für Pflegebedürftige)
- IGiB-StimMT (Umstrukturierung der Versorgung im ländlichen Raum)
- Rise-uP (verbesserte Schmerztherapie)
- PROMoting Quality (Qualitätssicherung nach Hüft-Endoprothese)
- STEP.De (Sporttherapie)
Gleichzeitig sieht man, warum Evidenz oft schwer ist: In mehreren Projekten waren die Fallzahlen wegen hoher Drop-outs in der Corona-Zeit nicht ausreichend, häufig gab es nur „tendenzielle“ Einsparungen, teils sogar höhere Kosten in der Interventionsgruppe. Trotzdem hatten laut BASYS alle 29 Projekte mit Umsetzungsempfehlung eine Outcome-Verbesserung, davon 5 mit signifikanter und 4 mit tendenzieller Kostenersparnis. Unter den Projekten mit tendenzieller Kostenersparnis sind z.B. auch diese zwei mit digitalem Schwerpunkt:
- Eric (multiprofessionelle, telemedizinische Visite)
- pAVK-TeGeCoach (telemetrisch gestütztes Gehtraining)
7. Fazit: Lohnt sich der Innovationsfonds für Hersteller medizinischer Software?
Der Innovationsfonds bietet Softwareherstellern eine Chance, außerhalb der klassischen Erstattungspfade an Finanzmittel zu gelangen und ihr Produkt in der Versorgungsrealität zu erproben. Besonders für innovative Lösungen, die einen Platz im Gesundheitssystem suchen, kann er als Katalysator dienen. Durch die Förderung entstehen Evidenz und Erfahrungen, die später den Eintritt in die Regelversorgung ebnen können, etwa über DiGA, Selektivverträge oder andere Erstattungswege.
Die Zwischenbilanz nach zehn Jahren unterstreicht das: Die 29 zur Umsetzung empfohlenen NVF-Projekte konnten eine Verbesserung der Versorgungsqualität nachweisen. Und die BASYS-Auswertung macht deutlich, dass die Einbindung der Industrie, insbesondere im Bereich digitaler Lösungen und KI, ausdrücklich gefordert wird. Wer als Softwarehersteller neue Versorgungsmodelle mitgestalten möchte, findet im Innovationsfonds also ein Instrument, das politisch gewollt ist.
Allerdings ist die Förderung kein einfacher oder kurzfristiger Weg. Zwischen 2016 und 2021 wurden über 2.000 Anträge eingereicht, knapp mehr als 500 Projekte bewilligt. Die Erfolgswahrscheinlichkeit lag bei grob 20 bis 25 Prozent. Hersteller sollten sich bewusst sein, dass erheblicher Aufwand ohne Erfolgsgarantie investiert wird. Hinzu kommt: Selbst bei erfolgreichem Projektabschluss gibt es keinen Automatismus für die Überführung in die Regelversorgung. Der Weg von der Förderung zur dauerhaften Erstattung erfordert auch nach Projektende aktives Engagement.
Wenn Sie einen Umsetzungspartner für Ihr Software- oder App-Vorhaben suchen, kontaktieren Sie uns gerne. Wir sind auf die Entwicklung und Zulassung digitaler Versorgungslösungen spezialisiert. QuickBird Medical hat Software-Produkte wie INTEGRATE-ATMP für Kunden entwickelt, die erfolgreich im Rahmen des Innovationsfonds gefördert wurden.
8. Weitere Informationen zum Innovationsfonds
Die Inhalte dieses Artikels basieren auf dem aktuellen Stand der öffentlichen Förderinformationen und Praxiserfahrungen mit dem Innovationsfonds. Da sich Rahmenbedingungen und Anforderungen regelmäßig weiterentwickeln, empfehlen wir, zusätzlich die offiziellen Unterlagen des Innovationsausschusses beim G-BA zu berücksichtigen.
Eine gute erste Anlaufstelle ist der Leitfaden zur Antragstellung für neue Versorgungsformen, den der G-BA regelmäßig aktualisiert: Zum Leitfaden
Auch aktuelle Präsentationen und Hinweise zur jeweils laufenden Förderbekanntmachung sind auf der offiziellen Website abrufbar: https://innovationsfonds.g-ba.de
Neben dem Innovationsfonds gibt es eine Reihe weiterer öffentlicher Förderungen, die sich gut für Digital-Health-Produkte eignen. Wir haben hierzu einen umfangreichen Leitfaden geschrieben, der verschiedene Typen von Förderungen erläutert und praxisnahe Ratschläge gibt, welche davon sich wirklich lohnen: Zum Whitepaper
The post Innovationsfonds: Förderung für digitale Gesundheitslösungen appeared first on QuickBird Medical.
]]>The post PECAN & DMD – Leitfaden für DiGA in Frankreich appeared first on QuickBird Medical.
]]>Was in Deutschland als „digitale Gesundheitsanwendung“ (DiGA) bezeichnet wird, heißt in Frankreich „Dispositifs Médicaux Numériques“ (DMN) und im internationalen Kontext „Digital Medical Device“ (DMD).
Für Hersteller entsteht damit ein strukturiertes Schnellverfahren, um digitale Medizinprodukte für einen zeitlich begrenzten Zeitraum über die gesetzliche Krankenversicherung in Frankreich erstatten zu lassen.
In diesem Beitrag liefern wir einen Leitfaden, der erklärt …
- wie PECAN funktioniert,
- inwiefern das Verfahren vom deutschen Erstattungsweg abweicht,
- welche Anforderungen gelten,
- und warum ein Transfer einer DiGA nach Frankreich zwar naheliegt, in der Praxis aber anspruchsvoll ist.
Überblick
- 1. Hintergrund: DiGA-Modell in Deutschland
- 2. Einführung: Erstattung von DMD in Frankreich: PECAN
- 3. Regulatorischer Rahmen des PECAN
- 4. Antragsverfahren des PECAN
- 5. Verfahren nach positiver PECAN-Entscheidung
- 6. Kostenerstattung von DMD
- 7. Klinische Bewertung im PECAN-Verfahren
- 8. Akteure und Zuständigkeiten
- 9. Stand 2026: DiGA-Verzeichnis von Frankreich
- 10. Vergleich: DiGA Fasttrack vs. DMD und PECAN
- 11. Von Deutschland nach Frankreich: DiGA zu PECAN
- 12. Hilfreiche Links
- 13. DiGA-Modelle in anderen Ländern
- 14. Fazit: Das DMD-Framework als DiGA mit Zusätzen
1. Hintergrund: DiGA-Modell in Deutschland
Seit 2019 gibt es in Deutschland mit dem DiGA-Fast-Track einen standardisierten Erstattungsweg für „Digitale Gesundheitsanwendungen“ (DiGA). Hersteller können beim Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) einen Antrag auf Aufnahme in das DiGA-Verzeichnis stellen. Im Rahmen des Fast-Tracks prüft das BfArM, ob die Anwendung die regulatorischen und formalen Anforderungen erfüllt (u. a. MDR/CE, Datenschutz/IT-Sicherheit) und ob ein positiver Versorgungseffekt nachgewiesen ist. Bei positiver Entscheidung wird die DiGA in das Verzeichnis aufgenommen und ist anschließend über die gesetzlichen Krankenkassen erstattungsfähig.
Der Fast-Track kennt dabei zwei mögliche Einstiegswege:
- Entweder erfolgt direkt eine dauerhafte Aufnahme, wenn der Nutzennachweis bereits vollständig vorliegt,
- oder eine vorläufige Aufnahme, wenn noch keine vollständige Studie zum positiven Versorgungseffekt existiert. In diesem Fall muss der Hersteller trotzdem ein Evaluationskonzept inklusive Evidenz aus einer Pilotstudie vorlegen. Bei positiver Bewertung wird die DiGA dann erstattet, während der Hersteller im sogenannten Erprobungsjahr die fehlenden Daten für eine vollständige Studie nachreicht. Erst nach erfolgreichem Abschluss kann die Anwendung dauerhaft im DiGA-Verzeichnis verbleiben.
Das französische Modell lehnt sich an dieses deutsche Modell zur Erstattung von digitalen Gesundheitsanwendungen an, hat aber gleichzeitig einige fundamentale Unterschiede.
2. Einführung: Erstattung von DMD in Frankreich: PECAN
PECAN steht für „Prise en Charge Anticipée Numérique“. Wörtlich: vorläufige digitale Kostenübernahme. Das Verfahren ermöglicht es Herstellern seit 2023, digitale Medizinprodukte mit vermutetem Innovationscharakter für maximal zwölf Monate vorläufig über die gesetzliche Krankenversicherung zu erstatten.
Frankreich hat mit PECAN vor allem die Idee der vorläufigen Erstattung aus dem DiGA-Fast-Track aufgegriffen. Der zentrale Unterschied zu Deutschland ist nämlich, dass der Übergang in die dauerhafte Erstattung nicht im selben Verfahren erfolgt. Die dauerhafte Erstattung muss anschließend über einen separaten regulären Weg (LPPR oder LATM) beantragt werden, der schon lange vor PECAN existierte.
Digitale Gesundheitsanwendungen werden in Frankreich zudem nicht als DiGA bezeichnet, sondern als „Dispositifs Médicaux Numériques“ (DMN) und im internationalen Kontext als „Digital Medical Devices“, kurz DMD.
3. Regulatorischer Rahmen des PECAN
Die rechtliche Grundlage des PECAN-Verfahrens ist Artikel L. 162-1-23 des französischen Sozialversicherungsrechts (Code de la Sécurité Sociale, CSS). Damit ist PECAN gesetzlich verankert und als vorläufiger Erstattungsmechanismus rechtlich verbindlich geregelt.
Zusätzlich stellt die französische Gesundheitsbehörde Haute Autorité de Santé (HAS) einen Leitfaden zur Verfügung, der Herstellern als praktisches Handbuch dienen soll. Er beschreibt nicht nur die wissenschaftlichen Anforderungen an die Nutzenvermutung, sondern konkretisiert auch die formalen Kriterien für technische Nachweise, insbesondere zu Interoperabilität und IT-Sicherheit. Ergänzend enthält das Dokument aktuelle Praxisbeispiele. Im Vergleich zum deutschen DiGA-Leitfaden fällt er mit rund 27 Seiten deutlich kompakter aus.
3.1 Anwendungsbereich
Der Anwendungsbereich umfasst zwei Kategorien digitaler Medizinprodukte:
- digitale Medizinprodukte mit therapeutischer Zweckbestimmung und
- digitale Medizinprodukte zur medizinischen Teleüberwachung.
Im Vergleich dazu ist der Anwendungsbereich des deutschen DiGA-Fast-Tracks enger gefasst und schließt keine Telemonitoring-Anwendungen ein.
3.2 Zentrale Prinzipien
Das PECAN-Verfahren folgt laut dem Leitfaden vier Prinzipien, die Hersteller kennen sollten:
- Fast-Track zur vorläufigen Erstattung:
PECAN ist ein gesondertes Verfahren, das der regulären Erstattung vorgelagert ist. - Zeitlich strikt begrenzt:
Die vorläufige Erstattung gilt maximal 12 Monate ab Entscheidung, eine Verlängerung ist ausgeschlossen. Eine weitere Kostenerstattung ist nur über einen separaten Antrag in der Regelversorgung (LPPR oder LATM) möglich. - Kumulative Förderkriterien:
Alle gesetzlichen Voraussetzungen müssen gleichzeitig erfüllt sein. - Technische Zertifizierung:
Zusätzlich zur Bewertung durch die Commission nationale d’évaluation des dispositifs médicaux et des technologies de santé (CNEDiMTS) ist eine Zertifizierung durch die Agence du Numérique en Santé (ANS) erforderlich. Die einzelnen Institutionen werden in Kapitel 8 nochmal detaillierter betrachtet.
3.3 Erstattungskriterien und Anforderungen für DMD in Frankreich
Damit ein DMD für das PECAN-Verfahren infrage kommt, müssen die folgenden Kriterien und Anforderungen erfüllt sein. Wichtig ist dabei: Die Bewertung erfolgt immer indikationsspezifisch. Ein Produkt kann also für eine Indikation PECAN-fähig sein, für eine andere nicht.
3.3.1 Erstattungskriterium 1: CE-Kennzeichnung nach MDR
Kurz gesagt: Ohne CE kein PECAN.
Wie auch in Deutschland gilt: Nicht jede Software ist eine digitale Gesundheitsanwendung, aber jede DMD ist ein Medizinprodukt und unterliegt der Medical Device Regulation (MDR).
Für PECAN bedeutet das:
- Das DMD muss eine gültige CE-Kennzeichnung nach MDR besitzen.
- Die beantragte Indikation muss mit der Indikation für die CE-Zertifizierung nach MDR übereinstimmen.
So wird geprüft:
- Maßgeblich ist die Indikation in der Gebrauchsanweisung.
- Weicht die im PECAN-Antrag genannte Indikation sprachlich oder inhaltlich davon ab, muss der Hersteller dies im Dossier klar begründen und erläutern.
Weitere Informationen zur Zulassung und Zertifizierung von Medizinprodukt-Software nach MDR finden Sie in unserem Leitfaden zum Thema.
3.3.2 Erstattungskriterium 2: Vermutete Innovation
Neben der CE-Kennzeichnung muss das DMD als mutmaßlich innovativ gelten. Entscheidend ist dabei, ob die Technologie:
- einen klinischen Nutzen erwarten lässt
oder - einen messbaren Fortschritt in der Organisation der Versorgung bietet.
Zwei Mindestanforderungen gelten dabei immer:
- Ein organisatorischer Fortschritt darf die Versorgungsqualität nicht beeinträchtigen.
- Für das digitale Medizinprodukt müssen laufende Studien vorliegen, die voraussichtlich genügend Daten liefern, um anschließend die dauerhafte Erstattung beantragen zu können.
Deutschland nutzt hierfür einen ähnlichen Bewertungsansatz: Statt „mutmaßlicher Innovation“ ist im DiGA-Fast-Track der „positive Versorgungseffekt“ entscheidend, der entweder über einen medizinischen Nutzen (mN) oder über eine patientenrelevante Struktur- und Verfahrensverbesserung (pSVV) nachgewiesen werden muss.
3.3.3 Erstattungskriterium 3: Technische Voraussetzungen
Parallel zur Bewertung durch die CNEDiMTS prüft die ANS die technischen Anforderungen.
Konkret muss das DMD:
- die Vorgaben zum Datenschutz erfüllen,
- die nationalen Interoperabilitäts- und IT-Sicherheitsstandards einhalten,
- einen standardisierten, interoperablen Datenexport ermöglichen,
- ggf. Schnittstellen zu Geräten zur Erfassung von Vitalparametern bieten.
Für die dauerhafte Erstattung ist zudem ein offizielles Interoperabilitätszertifikat erforderlich, u. a. im Zusammenspiel mit der französischen nationalen eHealth-ID (INS).
Auch mit dem DiGA-Fast-Track in Deutschland sind Datenschutz, Informationssicherheit und Interoperabilität zentrale Anforderungen.

Erstattungskriterien für PECAN im Vergleich zu DiGA
3.4 Ausschlussgründe
Eine vorläufige Erstattung ist nicht möglich, wenn:
- das DMD bereits eine PECAN-Erstattung für dieselbe Indikation hatte,
- eine PECAN bereits abgelehnt wurde
→ Ausnahme: Neue Daten bei zuvor unzureichender Evidenz, - eine behördliche Aussetzung oder ein Verbot besteht,
- PECAN mit anderen Erstattungsmechanismen kombiniert werden soll (z. B. LPPR, LATM, Innovationspauschale, Übergangserstattung).
4. Antragsverfahren des PECAN
Das Antragsverfahren besteht insgesamt aus drei Phasen.
- Einreichung & formale Prüfung des Dossiers
Der Hersteller reicht das Dossier bei den zuständigen Ministerien für Gesundheit und soziale Sicherheit ein, mit Kopie an die HAS.
In dieser ersten Phase wird geprüft:
- Ist das Dossier vollständig?
- Entspricht es den formalen Anforderungen der HAS?
Falls Unterlagen fehlen, erhält der Hersteller eine Nachforderung.
Frist: 30 Tage, um die fehlenden Informationen nachzureichen. Wer diese Frist verpasst, dessen Antrag wird als „zurückgezogen“ eingeordnet.
- Stellungnahme durch die CNEDiMTS
Bei vollständigem Dossier bewertet die CNEDiMTS die Förderfähigkeit. Im Fokus stehen Indikation und vermutete Innovation.
Die Bewertung erfolgt ohne Rückfragen oder Anhörungen der Antragsteller.
Bearbeitungszeit: maximal 60 Tage ab vollständigem Antrag.
Am Ende steht eine positive oder negative Stellungnahme, die:
- an die Ministerien,
- an den Hersteller,
- und an die betroffenen Fachgremien (CNP) geht und öffentlich auf der HAS-Website veröffentlicht wird.
- Entscheidung über die vorläufige Erstattung
Auf Basis der CNEDiMTS-Stellungnahme entscheiden die zuständigen Ministerien.
Frist: 30 Tage
Die Entscheidung wird per ministeriellem Erlass im „Journal officiel“ veröffentlicht und legt fest:
- ob das Produkt vorläufig erstattet wird,
- und zu welchen finanziellen Bedingungen.
5. Verfahren nach positiver PECAN-Entscheidung
Nach einer positiven PECAN-Entscheidung wird das digitale Medizinprodukt für maximal zwölf Monate vorläufig erstattet. In dieser Zeit muss der Hersteller den Übergang in die dauerhafte Erstattung vorbereiten. PECAN dient quasi als Brücke zur regulären Erstattung:
- Für digitale Medizinprodukte mit therapeutischer Zweckbestimmung ist der reguläre Erstattungsweg die Aufnahme in die Liste der erstattungsfähigen Produkte und Leistungen LPPR. Der Hersteller muss den Folgeantrag innerhalb von 6 Monaten nach der PECAN-Entscheidung einreichen. Auch hierfür stellt die HAS einen Leitfaden bereit.
- Für digitale Medizinprodukte zur medizinischen Teleüberwachung ist der reguläre Erstattungsweg die Aufnahme in die Liste der Aktivitäten der medizinischen Teleüberwachung LATM. Der Hersteller muss den Folgeantrag innerhalb von 9 Monaten nach der PECAN-Entscheidung einreichen. Auch hierfür stellt die HAS einen Leitfaden bereit.
Die untenstehende Abbildung fasst das PECAN-Verfahren und die nachfolgenden regulären Erstattungswege zusammen.

PECAN-Verfahren und anschließender Erstattungsweg der Regelversorgung
6. Kostenerstattung von DMD
Hier gehen wir auf die konkreten Regelungen zur Kostenerstattung von DMD im Rahmen des PECAN und danach ein.
6.1 Erstattungshöhe während des PECAN-Jahrs
Während des PECAN-Verfahrens werden Preise über staatlich definierte Pauschalen geregelt. Diese umfassen für:
Digitale Therapeutika (DTx)
- ein Erstpaket in Höhe von 435 €. Dieses kann einmalig pro Patienten abgerechnet werden. Es gilt für die effektive Nutzung des digitalen Medizinprodukts für maximal 3 Monate.
- Ein Folgepaket, das an das Erstpaket anschließt. Dieses wird anteilig entsprechend der vorgesehenen Verordnungsdauer und der tatsächlichen Nutzung abgerechnet und beträgt 38,30 € pro Monat.
- Die maximale finanzielle Entschädigung, bestehend aus den beiden zuvor genannten Pauschalbeträgen, beträgt 780 € pro Patienten und Jahr.
Weitere Informationen finden Sie im Absatz II des Artikels R. 162-117 des Sozialversicherungsgesetzbuches.
Für Telemonitoring-Lösungen
- erhält der technische Betreiber eine monatliche Pauschale von 50 € bis 91,67 € je nach nachgewiesenem Interesse.
- Die medizinische Überwachungsleistung wird separat vergütet (z. B. ärztlich oder pflegerisch).
Weitere Informationen finden Sie im Artikel L162-52 des Sozialversicherungsgesetzbuches.
6.2 Erstattung nach Ablauf des PECAN-Jahrs
Nach dem PECAN-Jahr endet die Pauschalvergütung. Für die dauerhafte Erstattung wird der Preis anschließend im jeweiligen Regelpfad über LPPR (für DTx) oder LATM (für Telemonitoring-Anwendungen) neu verhandelt, nutzbasiert und unter Einbindung des Comité économique des produits de santé (CEPS).
LPPR für digitale Gesundheitsanwendungen bzw. DTx
Im Regelpfad der LPPR erfolgt die Erstattung als Preis pro Einheit. Wichtig ist dabei, dass der tatsächliche Produktpreis höher sein kann als der Betrag, den die Krankenkasse erstattet. Die Differenz wird dann vom Patienten, seiner Zusatzkrankenversicherung (Mutuelle) oder einer anderen Organisation übernommen.
LATM für Telemonitoring
Im Regelpfad der LATM für Telemonitoring werden pro Patienten zwei Pauschalbeträge erstattet:
- ein Pauschalbetrag, der an den Fernüberwachungsbetreiber gezahlt wird, sowie
- ein Pauschalbetrag, der an den Betreiber gezahlt wird.
Die Sätze für die technischen Pakete werden dabei unter Berücksichtigung des organisatorischen oder klinischen Interesses festgelegt, das von der medizinischen Fernüberwachung zu erwarten ist. Das klinische Interesse wird anhand der Auswirkungen auf die Lebensqualität, die Morbidität oder die Mortalität bewertet.
7. Klinische Bewertung im PECAN-Verfahren
Die klinische Bewertung ist ein zentraler Bestandteil des PECAN-Verfahrens. Die CNEDiMTS prüft dabei, ob ein DMD als mutmaßlich innovativ gelten kann und ob die vorgelegten sowie laufenden Studien geeignet sind, kurzfristig die Grundlage für eine reguläre Erstattung über LPPR oder LATM zu schaffen. Maßgeblich sind dabei die Prinzipien der evidenzbasierten Medizin.
7.1 Evidenzbasiert und kontextabhängig
Die Bewertung erfolgt nicht losgelöst von der Versorgungspraxis. Die CNEDiMTS beurteilt die klinische Relevanz der Daten stets im Zusammenhang mit:
- der zugrunde liegenden Erkrankung,
- ihrer Epidemiologie,
- sowie der therapeutischen Strategie, in die das digitale Medizinprodukt eingebettet ist.
Entscheidend ist zudem, ob sich die Ergebnisse auf das französische Gesundheitssystem übertragen lassen. Bei begrenzten Rekrutierungsmöglichkeiten können internationale multizentrische Studien akzeptiert werden. Voraussetzung ist aber, dass ihre Übertragbarkeit auf das französische System nachvollziehbar begründet werden kann.
7.2 Datenbasis
Grundsätzlich stützt sich die CNEDiMTS bevorzugt auf produktspezifische klinische Daten. In bestimmten Fällen können jedoch auch nicht-produktspezifische Daten berücksichtigt werden, etwa zu technologisch vergleichbaren Lösungen oder früheren Versionen des Produkts.
Voraussetzung ist in diesen Fällen:
- eine schlüssige Begründung der technischen Äquivalenz,
- eine transparente Analyse möglicher Unterschiede,
- sowie der Nachweis, dass diese Unterschiede keinen relevanten Einfluss auf den klinischen oder technischen Effekt haben.
7.3 Klinischer Nutzen und Endpunkte
Der klinische Nutzen wird immer im Vergleich zu einer relevanten Vergleichsalternative bewertet. Dabei berücksichtigt die CNEDiMTS sowohl positive Effekte als auch potenzielle Risiken der Anwendung.
Beispielhafte Bewertungsdimensionen im PECAN-Leitfaden des CNEDiMTS sind:
- Lebensqualität,
- Morbidität oder Mortalität,
- Komplikationen,
- Reduktion ungeplanter Krankenhausaufenthalte.
Wichtig ist, dass die gewählten Endpunkte zur beanspruchten Indikation und zum Wirkversprechen des Produkts passen. In der Praxis zeigt sich, dass formal korrekte, aber inhaltlich unpassende Endpunkte häufig zu kritischen Bewertungen führen.
7.4 Organisatorischer Nutzen als Innovationsargument
Neben dem klinischen Nutzen kann auch ein Fortschritt in der Organisation der Versorgung zur Begründung der Innovationsannahme herangezogen werden. Das kann etwa durch neue Versorgungsprozesse oder eine verbesserte Zugänglichkeit der Versorgung, insbesondere im Bereich der Teleüberwachung, geschehen.
Die CNEDiMTS stellt jedoch fest, dass dieser Nutzen in Anträgen häufig nur beschreibend, aber nicht systematisch belegt wird. Empfohlen wird daher, organisatorische Effekte strukturiert darzustellen, z. B. anhand der beteiligten Akteure, veränderter Prozesse und geeigneter Bewertungskriterien.
8. Akteure und Zuständigkeiten
Für das PECAN-Verfahren sind vor allem die folgenden Akteure im französischen Gesundheitssystem relevant:
- CNEDiMTS („Commission nationale d’évaluation des dispositifs médicaux et des technologies de santé“ bzw. Nationale Kommission zur Bewertung von Medizinprodukten und Gesundheitstechnologien): Bewertet die medizinische und versorgungsbezogene Evidenz.
- ANS („Agence du Numérique en Santé“ bzw. Nationale Agentur für digitale Gesundheit): Prüft technische Anforderungen wie Datenschutz, Interoperabilität und IT-Sicherheit.
- Ministère chargé de la Santé & Ministère chargé de la Sécurité sociale (bzw. Ministerien für Gesundheit und soziale Sicherheit): Treffen die finale Entscheidung über die vorläufige Kostenübernahme und setzen diese per ministeriellem Erlass um, basierend auf den Stellungnahmen der CNEDiMTS und der ANS.
- HAS („Haute Autorité de Santé“ bzw. Französische Gesundheitsbehörde): Bietet u.a. sogenannte „frühe Beratungsgespräche“ an. Diese richten sich an Hersteller, deren Produkte sich noch in der klinischen Entwicklung befinden. Inhaltlich können dabei Fragen zur klinischen Evidenzstrategie besprochen werden, optional auch in Kombination mit gesundheitsökonomischen Themen (Effizienzbewertung). Wichtig: Die Gespräche sind optional, unverbindlich, vertraulich und kostenfrei. Sie gelten nicht als Bewertung und lassen keine Rückschlüsse auf die spätere Entscheidung der CNEDiMTS zu.
- G_NIUS („Guichet National de l’Innovation et des Usages en e-Santé“ bzw. Nationale Anlaufstelle für Innovation und Anwendungen im Bereich E-Health): Ist eine nationale Plattform zur Förderung von Innovationen im Bereich Digital Health. Sie unterstützt Entwickler und Hersteller insbesondere bei der Orientierung im regulatorischen Umfeld, der Identifikation von Finanzierungs- und Fördermöglichkeiten, sowie der Vernetzung mit relevanten Akteuren im französischen E-Health-Ökosystem. Ziel ist es, die Markteinführung neuer digitaler Technologien zu erleichtern und zu beschleunigen.
9. Stand 2026: DiGA-Verzeichnis von Frankreich
Obwohl das PECAN-Verfahren seit 2023 operativ verfügbar ist, spielt es in der Versorgungspraxis bislang nur eine untergeordnete Rolle. Anders als in Deutschland gibt es kein eigenständiges öffentliches DMD-Register. Digitale Medizinprodukte erscheinen nur indirekt in bestehenden Erstattungsstrukturen:
- LPPR (Liste des Produits et Prestations Remboursables): zentrales Erstattungsverzeichnis für alle Medizinprodukte, ohne Kennzeichnung digitaler Anwendungen
- LATM (Liste des Activités de Télésurveillance Médicale): Verzeichnis für erstattungsfähige Telemonitoring-Leistungen, ebenfalls ohne expliziten DMD-Status
Damit gibt es keine öffentliche Übersicht, welche DMD aktuell (vorläufig) erstattet werden.
Öffentlich nachvollziehbar ist bislang nur eine Anwendung, die tatsächlich über PECAN erstattet wurde:
Cureety TechCare
- Telemonitoring-Lösung für onkologische Patienten
- Aufnahme in PECAN: 2023
- kein Übergang in die dauerhafte Erstattung über die LATM
- Als wesentliche Gründe werden fehlende oder nicht ausreichend belastbare Nachweise zum langfristigen klinischen Nutzen und organisatorischen Mehrwert genannt.
10. Vergleich: DiGA Fasttrack vs. DMD und PECAN
Deutschland und Frankreich verfolgen das gleiche Ziel: digitale Medizinprodukte schneller in die Versorgung zu bringen. Der Unterschied liegt vor allem in Struktur, Transparenz und Preislogik.
- Erstattungslogik:
Sowohl das deutsche DiGA-Verfahren als auch das französische System sehen eine vorläufige Erstattung vor. In Deutschland sind die vorläufige und die dauerhafte Erstattung innerhalb desselben DiGA-Regelwerks verankert. In Frankreich ist die PECAN-Erstattung ausdrücklich als temporärer Einstieg konzipiert und auf zwölf Monate begrenzt. Eine dauerhafte Erstattung erfordert anschließend einen eigenständigen Antrag über LPPR oder LATM, also etablierte, nicht speziell auf digitale Anwendungen zugeschnittene Erstattungsmechanismen. - Erstattung:
In beiden Ländern übernehmen die gesetzlichen Krankenkassen die Kosten für die Erstattung. - CE-Zertifizierung:
In beiden Systemen ist die CE-Kennzeichnung nach MDR zwingend. - Klinische Evaluation:
Beide Verfahren arbeiten evidenzbasiert. DiGA zielt auf den positiven Versorgungseffekt, PECAN auf mutmaßliche Innovation plus belastbare Studienplanung. - Datenschutz & IT-Sicherheit:
Die Anforderungen sind in beiden Ländern vergleichbar hoch. Deutschland bündelt die Prüfung im DiGA-Verfahren. Frankreich trennt sie organisatorisch: klinisch über die CNEDiMTS, technisch über die ANS. - Risikoklassen:
In Deutschland ist der DiGA-Fast-Track auf Medizinprodukte der Risikoklassen I, IIa und IIb beschränkt. Produkte der Klasse III sind nicht inkludiert.
PECAN enthält keine formale Begrenzung der MDR-Risikoklasse. - Register & Transparenz:
In Deutschland gibt es mit dem DiGA-Verzeichnis ein eigenes, öffentliches Register ausschließlich für DiGA-Anwendungen. Ein vergleichbares, separates Verzeichnis für digitale Medizinprodukte existiert in Frankreich bislang nicht, wodurch ein transparenter Marktüberblick deutlich erschwert wird. - Preisbildung:
Im DiGA-Fast-Track können Hersteller den Preis während des Erprobungszeitraums unter Einhaltung der Höchstbeträge frei festlegen. Für die dauerhafte Listung müssen sie einen Vergütungsbetrag mit dem GKV-Spitzenverband verhandeln.
PECAN hingegen arbeitet in der vorläufigen Phase mit staatlich fixierten Pauschalen. Der endgültige Preis wird nach Ablauf des PECAN mit dem CEPS im Rahmen der regulären Erstattungswege verhandelt.

Unterschiede zwischen DMD und DiGA-Fast-Track
11. Von Deutschland nach Frankreich: DiGA zu PECAN
Nach dem Vergleich liegt der Gedanke nahe: Wenn eine DiGA in Deutschland funktioniert, warum nicht auch in Frankreich?
Auf dem Papier gibt es viele Parallelen: MDR-Basis, frühe Erstattung, Fokus auf Evidenz. Können deutsche DiGA-Hersteller also einfach mit den existierenden Produkten und Studien nach Frankreich gehen, um diesen neuen Markt zu erschließen?
In der Praxis scheint der Transfer leider nicht so einfach zu sein. Evidenz aus klinischen Studien in Deutschland wird in Frankreich ggf. nicht so einfach akzeptiert.
11.1 Beispiel: DiGA von „HelloBetter“ – Antrag auf PECAN-Erstattung
Das zeigt auch der Fall HelloBetter Insomnie. Als Hersteller von mehreren in Deutschland gelisteten DiGA reichte HelloBetter auch ein Dossier im PECAN-Verfahren ein.
Das Ergebnis war eine negative Stellungnahme der HAS und damit keine vorläufige Erstattung über PECAN. Als Begründung wurde angeführt, dass die vorgelegten Daten die erforderliche „Vermutung von Innovation“ nicht ausreichend belegen konnten. Damit sei kein ausreichender klinischer Nutzen bzw. Versorgungsfortschritt nachgewiesen worden.
Der Fall macht deutlich: DiGA-Evidenz aus Deutschland ist nicht automatisch PECAN-fähig. Unterschiede in Indikationsabgrenzung, Comparatoren, Versorgungslogik und Endpunkten spielen eine zentrale Rolle.
Stellungnahmen:
- Stellungnahmen der HAS finden Sie hier und hier
- Stellungnahmen des Herstellers „HelloBetter“ zu dieser Ablehnung finden Sie hier
11.2 Blick nach vorn: Deutsch-französische Annäherung
Trotz solcher Rückschläge ist die Richtung klar: Harmonisierung statt Insellösungen.
Der G-BA und die HAS arbeiten bereits an einer engeren Abstimmung, insbesondere bei:
- klinischen Evidenzanforderungen
- methodischen Standards
- technischer Bewertung digitaler Medizinprodukte
Ziel:
Hersteller sollen ihre Evidenz perspektivisch parallel für beide Märkte nutzen können.
12. Hilfreiche Links
Für weitere Informationen in Bezug auf die Erstattung von digitalen Gesundheitsanwendungen in Frankreich finden Sie hier eine Sammlung von wichtigen Links:
Links bezüglich PECAN-Verfahren:
- Rechtsgrundlage PECAN (CSS Art. L. 162-1-23): Der gesetzliche Rahmen für die vorläufige Kostenübernahme von DMD.
- PECAN-Leitfaden CNEDiMTS (HAS): Zentrales Dokument für Hersteller: Bewertungslogik, Anforderungen, Beispiele, typische Ablehnungsgründe.
- Arrêtés zur PECAN-Vergütung (DTx, Telemonitoring): Die Rechtsakte, die die Pauschalen/Parameter für die vorläufige Vergütung festlegen bzw. aktualisieren.
- PECAN-Übersicht (G_NIUS): Kurze, gut verständliche Zusammenfassung von PECAN inkl. Einordnung in die Erstattungslogik.
Links für Erstattung in der Regelversorgung (LPPR & LATM):
- HAS „Principes d’évaluation / DM pathway“ (2010 Guide): Überblick über Akteure und Erstattungswege rund um Medizinprodukte in Frankreich (v. a. MDR-Kontext/Marktüberwachung).
- LPPR/AMELI-Datenbank: Öffentliches Erstattungsumfeld für Medizinprodukte und Telemonitoring-Leistungen, ohne eigenes DMD-Register.
- LPPR-Leitfaden (HAS): Offizieller Leitfaden zur Antragstellung und Bewertung für die Aufnahme von Medizinprodukten in die LPPR.
- LPPR-Finanzierung (G_NIUS): Übersicht, wie Erstattung und Preislogik innerhalb der LPPR praktisch funktionieren.
- LATM-Leitfaden (HAS): Offizieller Leitfaden zur Einreichung eines Dossiers für Telemonitoring-Leistungen im LATM-Pfad.
- LATM-Finanzierung (G_NIUS): Erklärung der Vergütungslogik im Telemonitoring, inkl. Rollen, Pauschalen und Abrechnungsprinzip.
13. DiGA-Modelle in anderen Ländern
Immer mehr Länder planen einen standardisierten Prozess, um digitale Gesundheitsanwendungen in die Regelversorgung zu bringen. Neben Frankreich sollten Sie außerdem einen Blick in die folgenden Länder werfen:
- Belgien (Link zum Leitfaden für die Zulassung von DiGA in Belgien)
- Österreich (DiGA in Österreich: Zulassung digitaler Gesundheitsanwendungen (2026))
- Deutschland (Insider-Tipps für die Zulassung von DiGA in Deutschland)
- Schweiz (Erstattung von Digitalen Gesundheitsanwendungen (DiGA & dGA))
- Italien (Kommen DiGA bald nach Italien?)
- Erstattung von DiGA in der EU (Überblick über Zulassung von DiGA in allen Ländern der EU)
Außerdem gibt es eine breite Palette von öffentlichen Förderungen (siehe unser Praxisleitfaden zu Digital Health-Förderungen) und anderen Erstattungswegen (siehe unser Whitepaper zu Selektivverträgen) für Health-Software.
14. Fazit: Das DMD-Framework als DiGA mit Zusätzen
PECAN ist seit 2023 ein klar definiertes Verfahren, das Frankreich erstmals einen strukturierten Weg für die vorläufige Erstattung digitaler Medizinprodukte gibt.
Gleichzeitig zeigt die bisherige Praxis, dass PECAN aktuell noch kein verlässlicher Marktzugangspfad ist. Transparenz ist begrenzt, ein eigenes Register fehlt und öffentlich nachvollziehbare Erfolgsbeispiele scheint es kaum zu geben. Außerdem bleibt der Übergang in die dauerhafte Erstattung eine hohe Hürde, insbesondere wenn klinischer Nutzen oder organisatorischer Mehrwert nicht indikationsspezifisch und kurzfristig belastbar belegt werden können. Auch erfolgreiche DiGA-Evidenz aus Deutschland lässt sich nicht automatisch auf PECAN übertragen, wie der Fall von HelloBetter zeigt.
Für Hersteller heißt das: PECAN ist derzeit eher ein anspruchsvolles Übergangsinstrument als ein stabiler Erstattungskanal. Wer PECAN ernsthaft verfolgt, sollte auf eine belastbare Studienplanung achten und eine Strategie, die von Beginn an auf die dauerhafte Erstattung nach dem PECAN ausgerichtet ist.
Sie wollen eine digitale Anwendung auf den französischen Markt bringen?
Falls Sie die Zulassung einer digitalen Anwendung in Frankreich oder Deutschland planen, kontaktieren Sie uns gern. QuickBird Medical ist auf die auftragsbasierte Entwicklung und Zulassung von digitalen Gesundheitsanwendungen und Software-Medizinprodukten spezialisiert. Wir entwickeln Ihre Anwendung, lassen diese als Medizinprodukt nach MDR zu und führen Sie Schritt für Schritt durch das PECAN-Verfahren.
The post PECAN & DMD – Leitfaden für DiGA in Frankreich appeared first on QuickBird Medical.
]]>The post DiGA als Medizinprodukt (MDR): Anforderungen & Unterschiede appeared first on QuickBird Medical.
]]>Dieser Artikel klärt wichtige Fragen zum Verhältnis zwischen DiGA und Medizinprodukten:
- Muss eine DiGA ein Medizinprodukt sein?
- Welche Risikoklassen sind für DiGA erlaubt?
- Was ist der Zusammenhang zwischen DiGA-Status und MDR?
- Welche Zusatzanforderungen müssen DiGA erfüllen?
- Darf eine DiGA mit Hardware-Medizinprodukten kombiniert werden?
Der Artikel richtet sich an angehende DiGA-Hersteller und andere Fachkreise, die verstehen wollen, welche regulatorischen Anforderungen für digitale Gesundheitsanwendungen gelten. Wir bei QuickBird Medical waren bereits in die Entwicklung von über 15 DiGA auf Auftragsbasis involviert und geben in Fachartikeln unser Wissen weiter.
Inhaltsverzeichnis
- 1. Zusammenspiel: DiGA und Medizinprodukt
- 2. Welche MDR-Medizinprodukt-Klassen sind für DiGA erlaubt?
- 3. Medizinprodukt vs. DiGA: Zusatzanforderungen für DiGA
- 4. Anbindung von medizinischen Geräten & Hardware an DiGA
- 5. Unterschiede zwischen Medizinprodukt und DiGA
- 6. Fazit: Empfehlungen für Hersteller
1. Zusammenspiel: DiGA und Medizinprodukt
Mit dem Konzept der DiGA wurde ein strukturierter Pfad geschaffen, um patientenorientierte medizinische Software und Apps in die Erstattung der gesetzlichen Krankenversicherung in Deutschland zu bringen.
Voraussetzung dafür: Die Anwendung muss zunächst als Medizinprodukt nach der europäischen Medical Device Regulation (MDR) auf den Markt gebracht werden. Als Medizinprodukt gilt Software, die vom Hersteller für einen medizinischen Zweck bestimmt ist: etwa zur Diagnose, Therapie oder Überwachung von Krankheiten. Erst mit gültiger CE-Kennzeichnung kann ein Hersteller den DiGA-Antrag beim Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) stellen.
Zusätzlich zur Zulassung als Medizinprodukt, muss eine medizinische Anwendung aber auch eine breite Palette weiterer Anforderungen erfüllen, um in das DiGA-Verzeichnis aufgenommen zu werden.

Anforderungen von DiGA und Software-Medizinprodukten
1.1 Muss eine DiGA also ein Medizinprodukt sein?
Die Antwort ist eindeutig: Ja, jede DiGA muss ein Medizinprodukt sein.
Das ergibt sich direkt aus der gesetzlichen Definition. § 33a SGB V definiert DiGA als „Medizinprodukte niedriger Risikoklasse, deren Hauptfunktion wesentlich auf digitalen Technologien beruht“. Eine DiGA ist also keine Alternative zum Medizinprodukt, sondern eine spezielle Kategorie von Medizinprodukten mit zusätzlichen Anforderungen.
1.2 Was bedeutet das konkret für Hersteller?
Bevor ein Hersteller überhaupt einen Antrag auf Aufnahme in das DiGA-Verzeichnis beim BfArM stellen kann, muss das Konformitätsbewertungsverfahren nach MDR vollständig abgeschlossen sein. Die CE-Kennzeichnung ist dabei der Nachweis, dass das Produkt die grundlegenden Sicherheits- und Leistungsanforderungen erfüllt und in Europa in Verkehr gebracht werden darf.
Wichtig zu verstehen: Das BfArM prüft im DiGA-Verfahren nicht die MDR-Konformität selbst. Diese wird vorausgesetzt und durch die CE-Kennzeichnung belegt. Das BfArM prüft stattdessen die zusätzlichen Anforderungen der DiGAV, etwa den Nachweis positiver Versorgungseffekte, Datenschutz und Interoperabilität.
1.3 Wann ist meine Software ein Medizinprodukt?
Bevor also die Frage nach dem DiGA-Status relevant wird, müssen Hersteller zunächst klären, ob ihre Software überhaupt ein Medizinprodukt ist. Diese Entscheidung ist grundlegend für den gesamten regulatorischen Weg.
Ob eine Software als Medizinprodukt gilt, hängt primär von der medizinischen Zweckbestimmung ab, die der Hersteller festlegt. Software ist nur dann ein Medizinprodukt, wenn sie einen medizinischen Zweck verfolgt, also wenn sie wie bereits erwähnt entweder die Diagnose, Therapie oder Überwachung von Krankheiten unterstützt.
Die Abgrenzung ist nicht immer trivial. Eine App, die einfach nur Fitnessdaten trackt, ist in der Regel kein Medizinprodukt. Dieselbe App mit einer Funktion zur Erkennung von Herzrhythmusstörungen hingegen schon. Entscheidend ist, welchen Zweck der Hersteller für das Produkt deklariert und welche Funktionen tatsächlich angeboten werden.
1.4 Wann ist Software kein Medizinprodukt?
Nicht jede Gesundheits-App ist automatisch ein Medizinprodukt. Folgende Kategorien fallen z. B. in der Regel nicht unter die MDR:
- Reine Datenspeicherung oder -übertragung: Software, die Gesundheitsdaten nur speichert oder weiterleitet, ohne sie medizinisch zu verarbeiten oder zu interpretieren
- Administrative Funktionen: Terminplanung, Erinnerungsfunktionen ohne medizinischen Kontext, Praxisverwaltung
- Allgemeine Gesundheitsinformationen: Apps, die generische Informationen bereitstellen, ohne individuelle Empfehlungen zu geben
- Lifestyle- und Wellness-Anwendungen: Fitness-Tracker, Ernährungs-Apps oder Meditations-Apps ohne medizinischen Zweck
Unser ausführlicher Leitfaden „Ist Ihre Software ein Medizinprodukt?“ behandelt dieses Thema im Detail mit konkreten Entscheidungshilfen.
2. Welche MDR-Medizinprodukt-Klassen sind für DiGA erlaubt?
Nicht jedes Medizinprodukt kann eine DiGA werden. Der Gesetzgeber hat die DiGA-Fähigkeit auf bestimmte Risikoklassen beschränkt. Dies spiegelt die Idee wider, dass DiGA als niedrigschwelliges Versorgungsangebot mit entsprechend begrenztem Risikopotenzial konzipiert sind.
Die folgende Übersicht zeigt, welche Risikoklassen für DiGA zugelassen sind:
 (Klicken zum Vergrößern)
(Klicken zum Vergrößern)
Bei Medizinprodukten der Klasse I kann der Hersteller die Konformitätsbewertung selbst durchführen und die Konformitätserklärung ausstellen. Bei Klasse IIa und IIb ist hingegen die Einbeziehung einer Benannten Stelle erforderlich, die das Qualitätsmanagementsystem und die technische Dokumentation prüft.
2.1 Erweiterung durch das Digital-Gesetz (DigiG)
Eine wichtige Änderung trat mit dem Digital-Gesetz (DigiG) am 26. März 2024 in Kraft: Seitdem können auch Medizinprodukte der Risikoklasse IIb in das DiGA-Verzeichnis aufgenommen werden. Zuvor waren nur die Klassen I und IIa zugelassen.
Diese Erweiterung ermöglicht DiGA mit höherem Risikoprofil. Allerdings gelten für Klasse-IIb-DiGA besondere Anforderungen: Eine vorläufige Aufnahme zur Erprobung ist zum Beispiel nicht möglich. Hersteller müssen bereits bei Antragstellung die Ergebnisse einer Studie zum Nachweis eines medizinischen Nutzens vorlegen.
2.2 Wie bestimme ich die Medizinprodukt-Risikoklasse für meine DiGA?
Die korrekte Klassifizierung ist einer der ersten und wichtigsten Schritte bei der Entwicklung einer DiGA. Sie bestimmt nicht nur, ob eine Benannte Stelle einbezogen werden muss, sondern beeinflusst den gesamten regulatorischen Aufwand und die Kosten.
Die Klassifizierung von Software-Medizinprodukten erfolgt nach den Regeln in Anhang VIII der MDR. Für Software ist dabei Regel 11 besonders relevant. Diese Regel klassifiziert Software u. a. anhand des Risikos, das von den bereitgestellten Informationen für den Patienten ausgeht.
Die Klassifizierung hängt von mehreren Faktoren ab: der Art der Erkrankung (schwerwiegend vs. nicht-schwerwiegend), der Art der Entscheidungsunterstützung (Diagnose, Therapie, Überwachung) und der Reversibilität möglicher Schäden durch falsche Informationen.
Unser Artikel Klassifizierung von Software-Medizinprodukten erläutert die Klassifizierungsregeln im Detail mit praktischen Beispielen.
Praxistipp: Befassen Sie sich frühzeitig mit der Klassifizierung – idealerweise bereits in der Konzeptionsphase. Die Risikoklasse bestimmt den regulatorischen Aufwand, die Kosten und den Zeitrahmen bis zur Markteinführung maßgeblich.
3. Medizinprodukt vs. DiGA: Zusatzanforderungen für DiGA
Eine DiGA muss zwei regulatorische Ebenen erfüllen: zunächst die MDR-Anforderungen als Medizinprodukt und darüber hinaus die spezifischen Anforderungen der DiGA-Verordnung (DIGAV). Dieser Abschnitt gibt einen Überblick über beide Ebenen und zeigt, welche zusätzlichen Hürden DiGA-Hersteller im Vergleich zu „normalen“ Medizinprodukte-Herstellern nehmen müssen.
3.1 Anforderungen als Medizinprodukt (MDR)
Jede DiGA muss zunächst die grundlegenden MDR-Anforderungen erfüllen. Diese bilden das Fundament, auf dem die DiGA-spezifischen Anforderungen aufbauen. Die MDR verlangt ein umfassendes Qualitäts- und Dokumentationssystem.
Zu den zentralen Anforderungen gehören:
- Qualitätsmanagementsystem nach ISO 13485, das alle qualitätsrelevanten Prozesse dokumentiert und steuert
- Risikomanagement nach ISO 14971, das potenzielle Gefährdungen identifiziert und Maßnahmen zur Risikominimierung definiert
- Software-Lebenszyklus nach IEC 62304, der die Entwicklung und den Betrieb von Software strukturiert
- Gebrauchstauglichkeit nach IEC 62366-1, die sicherstellt, dass das Produkt sicher und effektiv bedient werden kann
- Klinische Bewertung, die den klinischen Nutzen und die Sicherheit des Produkts belegt
- Post-Market-Surveillance, die das Produkt nach dem Inverkehrbringen kontinuierlich überwacht
Diese Anforderungen gelten für alle Medizinprodukte. Dies ist unabhängig davon, ob sie zusätzlich als DiGA in die Erstattung gebracht werden sollen.
Unser Artikel Zulassung & Zertifizierung von Software-Medizinprodukten behandelt die MDR-Anforderungen ausführlich.
3.2 Zusatzanforderungen für DiGA (DiGAV)
Über die MDR-Medizinprodukt-Anforderungen hinaus definiert die DiGAV spezifische Kriterien, die eine DiGA erfüllen muss. Diese Anforderungen werden vom BfArM im Rahmen des Fast-Track-Verfahrens geprüft.
3.2.1 Positive Versorgungseffekte (pVE)
DiGA müssen einen positiven Versorgungseffekt nachweisen. Dieser kann entweder ein medizinischer Nutzen sein (z. B. Verbesserung des Gesundheitszustands) oder eine patientenrelevante Struktur- und Verfahrensverbesserung (z. B. bessere Therapieadhärenz, vereinfachter Zugang zur Versorgung).
Für die dauerhafte Aufnahme ins DiGA-Verzeichnis ist eine vergleichende Studie erforderlich, die den positiven Versorgungseffekt belegt. Bei vorläufiger Aufnahme erhält der Hersteller einen Erprobungszeitraum von bis zu 12 Monaten (in begründeten Ausnahmen bis zu 24 Monate), um diesen Nachweis zu erbringen. Auch für eine Aufnahme auf Erprobung muss aber ein umfangreiches Evaluationskonzept inklusive Pilotstudie vorgelegt werden.
Mehr dazu: Positive Versorgungseffekte nachweisen
3.2.2 Datenschutz
Die DiGAV stellt über die DSGVO hinaus besonders strenge Anforderungen an den Datenschutz. Es sind nur bestimmte, abschließend definierte Zwecke der Datenverarbeitung zulässig. Für die Verarbeitung von Gesundheitsdaten außerhalb Deutschlands gelten besondere Regelungen. Transparenz über Art und Umfang der Datenverarbeitung ist Pflicht.
Mehr dazu: DiGA Datenschutz-Anforderungen
3.2.3 Datensicherheit
Ein Informationssicherheitsmanagementsystem (ISMS) ist für DiGA-Hersteller verpflichtend und muss sogar nach ISO 27001 zertifiziert werden.
Zusätzlich müssen DiGA-Hersteller eine Zertifizierung nach BSI TR-03161 erlangen. Dies ist eine sehr anspruchsvolle Zertifizierung für die Informationssicherheit, die durch Prüfstellen und abschließend das BSI (Bundesamt für Sicherheit in der Informationstechnik) vergeben wird.
Mehr dazu: BSI TR-03161 für DiGA
3.2.4 Interoperabilität
DiGA müssen interoperable Standards nutzen, damit Gesundheitsdaten zwischen verschiedenen Systemen ausgetauscht werden können. Konkret bedeutet das: Die Möglichkeit, DiGA-Daten im menschenlesbaren und maschinenlesbaren Format exportieren zu können (durch Nutzung von FHIR-Profilen und dem MIO DiGA Toolkit) sowie die Anbindung an die elektronische Patientenakte (ePA).
Mehr dazu: Interoperabilität für DiGA
3.2.5 Qualitätsanforderungen
Die DiGAV definiert weitere Qualitätsanforderungen in verschiedenen Bereichen:
- Robustheit: Die DiGA muss stabil funktionieren und fehlertolerant sein
- Verbraucherschutz: Transparente Information, keine irreführende Werbung, Einhaltung des Heilmittelwerbegesetzes
- Nutzerfreundlichkeit: Barrierefreie Gestaltung, intuitive Bedienung
- Unterstützung der Leistungserbringenden: Sinnvolle Integration in bestehende Versorgungsprozesse
- Qualität medizinischer Inhalte: Evidenzbasierung, Aktualität, fachliche Korrektheit
- Patientensicherheit: Risikominimierende Maßnahmen, angemessene Warnhinweise
3.2.6 Anwendungsbegleitende Erfolgsmessung (AbEM)
Nach der Aufnahme in das DiGA-Verzeichnis müssen Sie kontinuierlich Daten zur Nutzung und Wirksamkeit ihrer DiGA erheben. Die anwendungsbegleitende Erfolgsmessung umfasst Nutzungsdaten, Patientenzufriedenheit (PGI-C-Skala) und perspektivisch indikationsspezifische PROMs. Diese Daten müssen regelmäßig an das BfArM berichtet werden.
3.3 Weiterführende Informationen
Mehr Informationen, welche Kriterien Sie einhalten müssen, um eine DiGA zu sein, finden Sie auch hier: DiGA Definition & Kriterien
4. Anbindung von medizinischen Geräten & Hardware an DiGA
Eine häufige Frage von Herstellern betrifft die Kombination von DiGA mit Hardware: Kann eine DiGA auch Sensoren, Wearables oder andere Medizingeräte einbinden? Die Antwort ist differenziert. Grundsätzlich ja, aber unter bestimmten Bedingungen.
DiGA sind primär Software-Medizinprodukte. Ihre Hauptfunktion muss wesentlich auf digitalen Technologien beruhen. Sie können jedoch mit Hardware-Medizinprodukten und anderen Systemen kommunizieren, Daten austauschen und diese verarbeiten – solange die digitale Hauptfunktion überwiegt.
4.1 Erstattungsfähige vs. nicht erstattungsfähige Hardware
Ein wichtiger Aspekt für Hersteller ist die Frage der Erstattungsfähigkeit. Nicht jede Hardware, die mit einer DiGA kombiniert wird, wird auch von der GKV erstattet.
Die folgende Übersicht zeigt exemplarische Beispiele, welche Hardware-Typen potenziell erstattet werden und welche nicht:
 (Klicken zum Vergrößern)
(Klicken zum Vergrößern)
Grundregel: Erstattungsfähig sind spezialisierte medizinische Geräte, die keine Gegenstände des täglichen Lebens darstellen. Gebrauchsgegenstände des täglichen Lebens, auch wenn sie medizinische Funktionen haben, werden nicht von der GKV bezahlt, können aber mit einer DiGA verwendet werden.
5. Unterschiede zwischen Medizinprodukt und DiGA
Nachdem wir die einzelnen Aspekte im Detail betrachtet haben, fassen wir einige wesentliche Unterschiede zwischen einem „normalen“ Medizinprodukt und einer DiGA zusammen.
 (Klicken zum Vergrößern)
(Klicken zum Vergrößern)
Diese Liste könnte noch sehr stark ausgeweitet werden und dient nur dem grundlegenden Verständnis.
6. Fazit: Empfehlungen für Hersteller
Der Weg zur DiGA führt immer über das Medizinprodukt. Wer eine digitale Gesundheitsanwendung entwickelt und in die GKV-Erstattung bringen möchte, muss beide regulatorischen Ebenen von Anfang an mitdenken.
Unsere Empfehlungen für Hersteller:
- Frühzeitig die regulatorische Strategie festlegen: Klären Sie zu Beginn, ob Ihr Produkt ein Medizinprodukt ist und ob der DiGA-Weg sinnvoll ist. Nicht jedes Medizinprodukt muss eine DiGA werden.
- MDR-Konformität als Basis sicherstellen: Ohne CE-Kennzeichnung keine DiGA. Planen Sie ausreichend Zeit für die Konformitätsbewertung ein. Das gilt insbesondere bei Klasse IIa und IIb, wo Benannte Stellen einbezogen werden müssen.
- DiGAV-Anforderungen parallel planen: Datenschutz, Datensicherheit und Interoperabilität sollten von Beginn an in die Produktentwicklung einfließen. Nachträgliche Anpassungen sind aufwendig und teuer.
- Studienplanung für den pVE-Nachweis frühzeitig starten: Insbesondere wenn Sie eine dauerhafte Aufnahme anstreben, beginnen Sie früh mit der Konzeption der Nachweisstudie.
- Beratung beim BfArM nutzen: Das BfArM bietet kostenpflichtige Beratungsgespräche an. Diese Investition lohnt sich, um Unsicherheiten im Vorfeld zu klären.
Sie planen eine DiGA? Wir entwickeln digitale Gesundheitsanwendungen im Auftrag für Unternehmen: von der Konzeption über die MDR-Zulassung bis zur Aufnahme ins DiGA-Verzeichnis. Wir übernehmen alle Schritte bis zur erfolgreichen Erstattung und danach.
Mehr Informationen zum Service zur DiGA-Entwicklung
The post DiGA als Medizinprodukt (MDR): Anforderungen & Unterschiede appeared first on QuickBird Medical.
]]>The post AI Act: Leitfaden für Medizinprodukt-Hersteller nach MDR (2026) appeared first on QuickBird Medical.
]]>- Aber ist die KI-Verordnung für mein Medizinprodukt überhaupt relevant?
- Welche zusätzlichen Anforderungen müssen speziell Medizinprodukt-Hersteller umsetzen?
- Und wie schaffe ich es, diese in mein bestehendes Qualitätsmanagementsystem zu integrieren?
Diese und weitere Fragen beantworten wir in diesem umfangreichen Leitfaden zum EU-AI Act. Wir fokussieren uns hierbei komplett auf die Auswirkungen der KI-Verordnung auf MDR-Medizinprodukte.
Inhalte des Leitfadens zur KI-Verordnung
- 1. Was ist der AI Act? (Stand 2026)
- 2. Ist der AI Act für mein Produkt relevant?
- 3. Die 3 Produktkategorien des AI Act
- 4. Anforderungen an Hochrisiko-KI-Systeme
- 5. Wie beeinflusst der AI Act die Zulassung meines Medizinprodukts?
- 6. Ab wann gilt der AI Act? Umsetzungs-Timeline
- 7. Fazit
1. Was ist der EU AI Act?
Der AI Act ist eine neue Verordnung, welche die Entwicklung und den Einsatz von künstlicher Intelligenz (KI, oder AI für Artificial Intelligence) in der EU regeln soll. Sie ist nicht auf Medizinprodukte beschränkt, sondern adressiert grundsätzlich jegliche Produkte, die unter die Definition eines KI-Systems fallen (siehe nächstes Kapitel “Ist der AI Act für mein Produkt relevant?”). Das übergeordnete Ziel der KI-Verordnung ist es, die Sicherheit und Grundrechte von Personen zu wahren. Die gesamte Verordnung finden Sie auf der Webseite der Webseite der Europäischen Union.
2. Ist der AI Act für mein Produkt relevant?
Der AI Act definiert KI-Systeme folgendermaßen:
Für die Zwecke dieser Verordnung bezeichnet der Ausdruck „KI-System“ ein maschinengestütztes System, das für einen in unterschiedlichem Grade autonomen Betrieb ausgelegt ist und das nach seiner Betriebsaufnahme anpassungsfähig sein kann und das aus den erhaltenen Eingaben für explizite oder implizite Ziele ableitet, wie Ausgaben wie etwa Vorhersagen, Inhalte, Empfehlungen oder Entscheidungen erstellt werden, die physische oder virtuelle Umgebungen beeinflussen können;
Zusammengefasst ist ein KI-System demnach eine Software, die aus den erhaltenen Inputs ableitet, wie ein bestimmter Output erzeugt werden kann. Zudem kann das System (aber muss nicht) autonom arbeiten und laufend weiterlernen. Unterm Strich ist damit zwar noch nicht 100% klar, welche Systeme davon genau exkludiert sind, aber da die meisten Produkte mit KI unter Machine Learning fallen, sind sie sehr eindeutig von dieser Definition erfasst.
Die MDCG hat ein Dokument veröffentlicht, welches das Zusammenspiel zwischen der MDR und dem AI Act verdeutlicht: MDCG 2025-6. Dieses Dokument ist speziell für Hersteller von Software-Medizinprodukten interessant. Dort werden Produkte, die sowohl der MDR, als auch dem AI Act unterliegen, als „Medical Device Artificial Intelligence (MDAI)“ definiert.
3. Die 3 Produktkategorien des AI Act
Gelten für alle KI-Medizinprodukte (MDAI) dieselben Anforderungen? Die kurze Antwort lautet: Nein. Ähnlich wie die MDR unterteilt auch der EU AI Act Produkte in verschiedene Klassen:
- Verbotene KI Praktiken
- Hochrisiko-KI-Systeme
- Andere KI-Systeme (mit geringem Risiko)
In welche Klasse fällt mein KI- bzw. Software-Medizinprodukt? Und was hat das für mich als Hersteller zu bedeuten? 
Entscheidungsbaum: In welche KI-Risikoklasse fällt mein KI- bzw Software-Mizinprodukt (MDAI)?
3.1 Produktkategorie 1: Verbotene KI-Praktiken
Auf das Wesentliche reduziert lässt sich sagen, dass vor allem solche Praktiken verboten sind, die zum Ziel haben, Personen zu schaden – und zwar durch unterschwellige Beeinflussung oder das Ausnutzen bestimmter Schwächen oder Schutzbedürftigkeiten (z.B. Behinderungen, Alter). Artikel 5 der KI-Verordnung listet noch eine Reihe weiterer verbotener Praktiken, die aber in der Regel für Medizinprodukte nicht relevant sind.
Fällt mein Medizinprodukt in diese Kategorie? Das Ziel, Personen zu schaden, widerspricht üblicherweise den Zielen eines Medizinprodukts, weshalb Sie grundsätzlich davon ausgehen können, dass Ihre KI nicht in die Kategorie der “verbotenen Praktiken” fällt. Dennoch sollten Sie sich die Kriterien in Artikel 5 genau ansehen, um sicherzugehen.
Welche Anforderungen muss ich bei „Verbotenen KI-Praktiken“ beachten? Das Produkt darf nicht auf den Markt gebracht werden.
3.2 Produktkategorie 2: Hochrisiko-KI-Systeme
In diese Kategorie fallen Produkte, die ein hohes Risiko für die Gesundheit und Sicherheit, oder die Grundrechte natürlicher Personen darstellen.
Fällt mein Medizinprodukt in diese Kategorie? Vor allem bei den Begriffen “Gesundheit” und “Sicherheit” liegt der Gedanke an “Medizinprodukte” nicht mehr fern, zumal auch die MDR einen starken Fokus auf die Sicherheit, der von ihr geregelten Produkte legt.
Gemäß MDCG 2025-6 ist ein Medizinprodukt dann ein Hochrisiko-KI-System, wenn folgende Kriterien erfüllt sind:
- Das KI-System ist selbst ein Medizinprodukt oder eine Sicherheitskomponente davon
- Es ist eine benannte Stelle in die Konformitätsbewertung nach MDR eingebunden
Wenn in das Konformitätsbewertungsverfahren Ihres Produkts eine benannte Stelle involviert ist (z.B., weil es in Risikoklasse IIa oder höher fällt), ist Ihr Medizinprodukt automatisch ein Hochrisiko-KI-System gemäß KI-Verordnung (siehe Entscheidungsbaum in der Abbildung oben). Die benannte Stelle hat dann nicht nur die Anforderungen der MDR, sondern auch jene des AI Act im Zuge der Konformitätsbewertung zu prüfen. Wenn aber keine benannte Stelle bei der Zulassung Ihres Produkts involviert ist (z.B., weil das Produkt in Risikoklasse I nach MDR fällt, oder es sich gar nicht um ein Medizinprodukt handelt), müssen Sie vor allem Anhang III des AI Acts prüfen. Dieser enthält eine Liste von Systemen, die als Hochrisiko-KI-Systeme gelten. Die meisten Medizinprodukte werden sich aber nicht in dieser Liste finden lassen, weshalb wir davon ausgehen, dass viele Medizinprodukte der Risikoklasse I nach MDR keine Hochrisiko-KI-Systeme sind.
Diese Einschätzung bestätigt auch die MDCG 2025-6, in welcher die untenstehende Tabelle aufgeführt ist:
 Einordnung von Hochrisiko-KI-Systemen nach MDR
Einordnung von Hochrisiko-KI-Systemen nach MDR
Welche Anforderungen muss ich beachten? Wenn Ihr Produkt in die Kategorie der Hochrisiko-KI-Systeme fällt, hat die KI-Verordnung möglicherweise starke Auswirkungen auf Ihre Organisation und Ihr Medizinprodukt. Die Inhalte des EU AI Acts definieren nämlich speziell Anforderungen an die Hersteller solcher Systeme und die Systeme selbst. Einen Überblick über die Anforderungen geben wir Ihnen weiter unten im Kapitel “Anforderungen an Hochrisiko-KI-Systeme”.
3.3 Produktkategorie 3: Andere KI-Systeme (mit geringem Risiko)
Alle KI-Produkte, die weder den verbotenen KI-Praktiken noch den Hochrisiko-KI-Systemen zugeordnet werden können, fallen in diese Kategorie. Es handelt sich dabei grob gesagt um Systeme, von denen keine nennenswerten Risiken für die Sicherheit und Grundrechte von Personen ausgehen. Fällt mein Medizinprodukt in diese Kategorie? Wenn Ihr Produkt die folgenden Kriterien erfüllt, fällt es vermutlich in diese Kategorie:
- Es handelt sich um ein Produkt der Risikoklasse I nach MDR (es ist keine benannte Stelle in die Konformitätsbewertung involviert)
- Es wurde ausgeschlossen, dass es sich um ein Hochrisiko-KI-System handelt
- Es wurde ausgeschlossen, dass es sich um verbotene KI-Praktiken handelt
Welche Anforderungen muss ich beachten? Im Vergleich zu Hochrisiko-KI-Systeme kommen Produkte in dieser Kategorie wirklich gut weg. Im Grunde findet sich im gesamten AI Act nur eine Anforderung an solche Systeme: Transparenzpflichten. Und diese gelten auch nur für solche Systeme, die mit Menschen interagieren, mediale Inhalte erzeugen, oder zur Emotionserkennung dienen. Grob gesagt müssen Hersteller solcher Produkte sicherstellen, dass Nutzer wissen, dass sie es mit KI zu tun haben, und dass es sich bei den erzeugten Inhalten um keine echten Bilder, Videos, etc. handelt (z.B. Deepfake). KI-Systeme dieser Kategorie müssen in keiner gesonderten Datenbank angemeldet werden und sie bedürfen keiner Konformitätserklärung. Wenn Ihr Medizinprodukt also in diese Kategorie fällt, sind die Aufwände für die Konformität mit dem AI Act für Sie vermutlich überschaubar.
3.4 Zusatzkategorie: KI-Modelle mit allgemeinem Verwendungszweck
Unabhängig von der Risiko-Einstufung können bestimmte KI-Modelle als „KI-Modell mit allgemeinem Verwendungszweck“ (Englisch: „General-purpose AI model“) gelten. Das sind Modelle, die für verschiedene Zwecke verwendet werden können.
Da der Zweck von Medizinprodukten aber meist sehr genau definiert ist, wird diese Kategorie in der Praxis nur für wenige Hersteller relevant sein. Der Vollständigkeit halber möchten wir sie an dieser Stelle aber trotzdem nennen.
Falls Sie ein KI-Modell entwickeln, das über die Anwendung in einem konkreten Produkt hinausgeht, sollten Sie sich mit dieser Kategorie näher beschäftigen.
Die genauen Anforderungen an solche Modelle sind in Kapitel V des AI Act definiert.
4. KI-Verordnung – Anforderungen an Hochrisiko-KI-Systeme und deren Hersteller
Die genauen Anforderungen an Hochrisiko-KI-Systeme und ihre Hersteller sind in Kapitel III Abschnitt 2 und Abschnitt 3 des AI Act spezifiziert. Wir gehen an dieser Stelle davon aus, dass Sie bereits die Anforderungen der MDR erfüllen. Somit gehen wir hier vor allem auf die Punkte ein, die über die Anforderungen der MDR hinausgehen. Erfahren Sie mehr über die Anforderungen der MDR an KI-Medizinprodukte in diesem Artikel: Zulassung & Zertifizierung von Software-Medizinprodukten (MDR)
Die KI-Verordnung definiert die Pflichten der Anbieter von Hochrisiko-KI-Systemen wie folgt (Artikel 16). Sie müssen …
a) sicherstellen, dass ihre Hochrisiko-KI-Systeme die in Abschnitt 2 festgelegten Anforderungen erfüllen;
b) auf dem Hochrisiko-KI-System oder, falls dies nicht möglich ist, auf seiner Verpackung oder in der beigefügten Dokumentation ihren Namen, ihren eingetragenen Handelsnamen bzw. ihre eingetragene Handelsmarke und ihre Kontaktanschrift angeben;
c) über ein Qualitätsmanagementsystem verfügen, das Artikel 17 entspricht;
d) die in Artikel 18 genannte Dokumentation aufbewahren;
e) die von ihren Hochrisiko-KI-Systemen automatisch erzeugten Protokolle gemäß Artikel 19 aufbewahren, wenn diese ihrer Kontrolle unterliegen;
f) sicherstellen, dass das Hochrisiko-KI-System dem betreffenden Konformitätsbewertungsverfahren gemäß Artikel 43 unterzogen wird, bevor es in Verkehr gebracht oder in Betrieb genommen wird;
g) eine EU-Konformitätserklärung gemäß Artikel 47 ausstellen;
h) die CE-Kennzeichnung an das Hochrisiko-KI-System oder, falls dies nicht möglich ist, auf seiner Verpackung oder in der beigefügten Dokumentation anbringen, um Konformität mit dieser Verordnung gemäß Artikel 48 anzuzeigen;
i) den in Artikel 49 Absatz 1 genannten Registrierungspflichten nachkommen;
j) die erforderlichen Korrekturmaßnahmen ergreifen und die gemäß Artikel 20 erforderlichen Informationen bereitstellen;
k) auf begründete Anfrage einer zuständigen nationalen Behörde nachweisen, dass das Hochrisiko-KI-System die Anforderungen in Abschnitt 2 erfüllt;
l) sicherstellen, dass das Hochrisiko-KI-System die Barrierefreiheitsanforderungen gemäß den Richtlinien (EU) 2016/2102 und (EU) 2019/882 erfüllt.
Keine Sorge, wir lassen Sie mit dieser Liste nicht allein. Im Folgenden wollen wir deshalb beleuchten, was das für Sie als Hersteller eines KI-Medizinprodukts bedeutet. Wir gehen dabei auf die wichtigsten Punkte ein, die zu einer Anpassung Ihres Managementsystems und/oder des Medizinprodukts führen.

Die Anforderungen des EU-AI Act und der MDR überschneiden sich in zahlreichen Punkten.
4.1 AI Act – Anforderungen an Ihr Qualitätsmanagementsystem
Ein Medizinprodukthersteller wird von der MDR bereits dazu verpflichtet, ein Qualitätsmanagementsystem zu etablieren. In den meisten Fällen wird dabei unter anderem die ISO 13485 und – wenn es um Software geht – die IEC 62304 umgesetzt. Auf den ersten Blick sieht es so aus, als hätten Sie damit auch die Anforderung des AI Acts erfüllt. Auf den zweiten Blick wird aber deutlich, dass dies nur teilweise stimmt. Aber keine Angst, der Großteil Ihres Systems kann bleiben, wie er ist. Sie müssen lediglich ein paar Anpassungen und Erweiterungen vornehmen. Die Anforderungen aus Artikel 17 des EU AI Acts finden Sie in der untenstehenden Tabelle aufgelistet. Zusätzlich zu den genauen Anforderungen der Verordnung haben wir die konkreten Maßnahmen für Sie definiert, die Sie umsetzen müssen, um die entsprechende Konformität Ihres Qualitätsmanagementsystems herzustellen. Das Qualitätsmanagementsystem muss mindestens die folgenden Aspekte umfassen:
(Hinweis: Unsere Kommentare sind nicht als absolute Wahrheiten zu verstehen, sondern sollen lediglich etwaige Gemeinsamkeiten und Unterschiede zwischen dem AI Act und Ihrem bestehenden System verdeutlichen. Prüfen Sie daher für jeden Punkt, inwiefern Sie Maßnahmen ergreifen müssen.)
| Wortlaut aus dem AI Act | Kommentar zur Umsetzung |
|---|---|
| a) ein Konzept zur Einhaltung der Regulierungsvorschriften, was die Einhaltung der Konformitätsbewertungsverfahren und der Verfahren für das Management von Änderungen an dem Hochrisiko-KI-System miteinschließt; | Da Sie als Medizinprodukt-Hersteller bereits ein Konzept zur Einhaltung der MDR, DSGVO und anderer regulatorischer Vorgaben haben, sollten Sie dieses einfach um den AI Act erweitern. |
| b) Techniken, Verfahren und systematische Maßnahmen für den Entwurf, die Entwurfskontrolle und die Entwurfsprüfung des Hochrisiko-KI-Systems; | Dieser Punkt könnte bereits durch die ISO 13485 und IEC 62304 umgesetzt sein. |
| c) Techniken, Verfahren und systematische Maßnahmen für die Entwicklung, Qualitätskontrolle und Qualitätssicherung des Hochrisiko-KI-Systems; | Dieser Punkt sollte durch die ISO 13485 und IEC 62304 bereits umgesetzt sein. |
| d) Untersuchungs-, Test- und Validierungsverfahren, die vor, während und nach der Entwicklung des Hochrisiko-KI-Systems durchzuführen sind, und die Häufigkeit der Durchführung; | Auch dieses Thema findet sich in der ISO 13485 wieder und als Medizinprodukt-Hersteller verfügen Sie bereits über entsprechende Prozesse. Prüfen Sie aber, ob Ihre Verfahren für das KI-System angemessen sind. |
| e) die technischen Spezifikationen und Normen, die anzuwenden sind und, falls die einschlägigen Normen nicht vollständig angewandt werden oder sie nicht alle relevanten Anforderungen gemäß Abschnitt 2 abdecken, die Mittel, mit denen gewährleistet werden soll, dass das Hochrisiko-KI-System diese Anforderungen erfüllt; | Harmonisierte Normen zur Umsetzung des AI Act werden aber im laufe der Zeit von der Europäischen Kommission festgelegt (Stand vom 20. Januar 2026). Dies wird vermutlich ähnlich aussehen, wie unter der MDR, deren harmonisierte Normen in einem Durchführungsbeschluss veröffentlicht sind und laufend erweitert werden. |
| f) Systeme und Verfahren für das Datenmanagement, einschließlich Datengewinnung, Datenerhebung, Datenanalyse, Datenkennzeichnung, Datenspeicherung, Datenfilterung, Datenauswertung, Datenaggregation, Vorratsdatenspeicherung und sonstiger Vorgänge in Bezug auf die Daten, die im Vorfeld und für die Zwecke des Inverkehrbringens oder der Inbetriebnahme von Hochrisiko-KI-Systemen durchgeführt werden; | Diese Verfahren müssen vermutlich neu etabliert werden, da sie nicht explizit von der MDR, oder den damit assoziierten Normen gefordert werden. Mehr dazu finden Sie weiter unten im Kapitel “Daten und Daten-Governance”. |
| g) das in Artikel 9 genannte Risikomanagementsystem; | Dieser Punkt sollte zum Großteil bereits durch die MDR (bzw. ISO 14971) abgedeckt sein. Prüfen Sie, inwieweit Ihr Risikomanagement die Anforderungen des AI Act schon erfüllt (siehe folgende Kapitel). |
| h) die Einrichtung, Anwendung und Aufrechterhaltung eines Systems zur Beobachtung nach dem Inverkehrbringen gemäß Artikel 72; | Dieser Punkt ist zum Großteil bereits durch die MDR (Post-Market Surveillance) abgedeckt. Prüfen Sie, inwieweit Ihr System zur Beobachtung nach dem Inverkehrbringen die Anforderungen des AI Act schon erfüllt (siehe folgende Kapitel). |
| i) Verfahren zur Meldung eines schwerwiegenden Vorfalls gemäß Artikel 73; | Dieser Punkt ist zum Großteil bereits durch die MDR (Vigilanz) abgedeckt. Für Medizinprodukte gibt es hier sogar eine Sonderregelung. Dadurch, dass Medizinprodukt-Hersteller bereits über ein solches System verfügen, fordert die KI-Verordnung lediglich, dass die neuen Anforderungen in dieses System integriert werden. Inwiefern diese überhaupt zu Änderungen an Ihrem System führen, müssen Sie prüfen. Es kann gut sein, dass hier wenig, bis gar kein Aufwand entsteht. |
| j) die Handhabung der Kommunikation mit zuständigen nationalen Behörden, anderen einschlägigen Behörden, auch Behörden, die den Zugang zu Daten gewähren oder erleichtern, notifizierten Stellen, anderen Akteuren, Kunden oder sonstigen interessierten Kreisen; | Die Kommunikation mit verschiedenen Stakeholdern müssen Sie durch die ISO 13485 bereits regeln. Erweitern Sie diese Liste also einfach um etwaige neue Behörden, oder Akteure, um Konformität mit diesem Punkt zu erlangen. Dies könnte beispielsweise dann relevant werden, wenn Sie Daten eines KI-Reallabors nutzen wollen. |
| k) Systeme und Verfahren für die Aufzeichnung sämtlicher einschlägigen Dokumentation und Informationen; | Diesen Punkt sollten Sie durch Ihr Qualitätsmanagementsystem schon umgesetzt haben. Hier geht es darum, Prozesse zu etablieren, die die notwendigen Outputs (z.B. technische Dokumentation) generieren. |
| l) Ressourcenmanagement, einschließlich Maßnahmen im Hinblick auf die Versorgungssicherheit; | Diesen Punkt sollten Sie durch Ihr Qualitätsmanagementsystem nach ISO 13485 bereits umgesetzt haben. |
| m) einen Rechenschaftsrahmen, der die Verantwortlichkeiten der Leitung und des sonstigen Personals in Bezug auf alle in diesem Absatz aufgeführten Aspekte regelt. | Dies Verantwortlichkeit können Sie auf Ihre PRRC nach MDR übertragen, oder eine neue Rolle dafür definieren. |
4.1.1 Risikomanagement
Als Medizinprodukt-Hersteller verfügen Sie bereits über ein Risikomanagementsystem und sind ggf. von verschiedenen Gesetzen zu Risikomanagement-Aktivitäten verpflichtet (z.B. MDR, DSGVO). Die Anforderungen an das Risikomanagementsystem sind aber in MDR und AI Act nicht deckungsgleich! Die wohl fundamentalste Unterschied ist, dass Sie nicht nur Risiken in Bezug auf die Sicherheit, sondern auch auf die Grundrechte von Personen berücksichtigen müssen. Teilweise kann dies durch die Einhaltung anderer Verordnungen (z.B. DSGVO) zwar umgesetzt sein, neben der körperlichen Unversehrtheit und dem Schutz personenbezogener Daten können aber auch andere Rechte durch ein KI-System verletzt werden. Werfen Sie daher auf jeden Fall einen Blick in die Grundrechte-Charta der Europäischen Union. Zudem müssen Hochrisiko-KI-Systeme mit geeigneten Verfahren regelmäßig getestet werden (dies gilt vermutlich vor allem für kontinuierlich weiterlernende Systeme, die unter der MDR nur in Ausnahmefällen zulassungsfähig sind). Dieses Testen ist auch erforderlich, um die geeignetsten Risikomanagementmaßnahmen zu ermitteln. Die ermittelten Risiken sind dabei so weit wie „technisch möglich“ und bis zu einem „vertretbar beurteilbaren“ Grad gesenkt werden.
4.1.2 System zur Beobachtung nach dem Inverkehrbringen
Durch die MDR müssen Sie bereits die Post-Market Surveillance Ihres Medizinprodukts sicherstellen. Darauf wird in der KI-Verordnung auch explizit eingegangen. Wenn Sie nämlich bereits ein solches System nach MDR etabliert haben, müssen Sie prüfen, ob dieses auch die folgende Anforderung erfüllt: Das System muss es Ihnen ermöglichen, die Konformität des Produkts fortlaufend bewerten zu können.
4.1.3 System zur Meldung schwerwiegender Vorfälle und Fehlfunktionen
Durch Ihr bereits etabliertes Vigilanz-System nach MDR gibt es in diesem Bereich wenig umzusetzen. Der AI Act hat hier sogar eine Sonderregel für Medizinprodukte. Dadurch, dass bereits eine Verpflichtung zur Meldung sicherheitsrelevanter Vorfälle besteht, müssen Sie Ihr System lediglich um die Meldung von “Verstößen gegen die Bestimmungen des Unionsrechts zum Schutz der Grundrechte” erweitern.
4.1.4 Daten und Daten-Governance
Hier werden Sie als Hersteller dazu verpflichtet, geeignete Prozesse zu etablieren, welche Folgendes regeln:
- Konzeptionelle Entscheidungen (z.B. Wahl des Modelltyps, Festlegung relevanter Features)
- Datenerfassung (z.B. Datenquellen)
- Datenaufbereitungsvorgänge (z.B. Labelling, Bereinigung, Aggregierung)
- Aufstellung relevanter Annahmen (z.B. Beziehung zwischen Features, potenzielle Einflussfaktoren)
- Vorherige Bewertung der Verfügbarkeit, Menge und Eignung der benötigten Datensätze
- Untersuchung im Hinblick auf mögliche Verzerrungen/Bias und geeignete Maßnahmen zu derer Erkennung, Verhinderung und Abschwächung
- Ermittlung möglicher Datenlücken, Mängel und Maßnahmen, diese zu beheben
Grundsätzlich stellt dieses Kapitel auch konkrete Anforderungen an Ihre Trainings-, Validierungs- und Testdaten. Diese Anforderungen sollten Sie direkt in Ihre Prozesse integrieren. Tipp: Berücksichtigen Sie bei der Erstellung dieser Prozesse auch die Anforderungen an die technische Dokumentation in Anhang IV und Artikel 11 der KI-Verordnung. Dadurch stellen Sie sicher, dass Ihre Verfahren auch die notwendigen Outputs generieren.
4.1.5 Aufbewahrungspflichten
Wenn Sie die Aufbewahrungspflichten der MDR einhalten, erfüllen Sie vermutlich auch jene des AI Acts. Dort ist definiert, dass Sie Ihre Dokumentation für einen Zeitraum von 10 Jahren nach dem Inverkehrbringen des KI-Systems aufbewahren müssen. Zu dieser Dokumentation zählt laut Artikel 18 der KI-Verordnung die technische Dokumentation, das Qualitätsmanagementsystem, von benannten Stellen freigegebene Änderungen und ausgestellte Dokumente, sowie die Konformitätserklärung. Vom Produkt automatisch erzeugte Protokolle müssen 6 Monate lang aufbewahrt werden.
4.2 AI Act – Anforderungen an die technische Dokumentation
„Technische Dokumentation“ ist ein Begriff, der Ihnen sicherlich bereits aus der MDR bekannt ist. Eine technische Dokumentation müssen Sie für Ihr Medizinprodukt ohnehin erstellen. Tatsächlich gibt es auch hier viele Parallelen und Sie werden die meisten Punkte bereits durch die Einhaltung der MDR umgesetzt haben. Darunter zählen beispielsweise die Produktbeschreibung, eine Konformitätserklärung und die Methoden, die bei der Produktentwicklung angewandt wurden. Dies gilt aber nicht in jedem Fall! Besonders für Software-Systeme der Software-Sicherheitsklasse A nach IEC 62304 könnten sich in Bezug auf die technische Dokumentation einige Besonderheiten ergeben. Die KI-Verordnung verpflichtet Sie z.B. dazu, eine Systemarchitektur und die Definition von Schnittstellen zu anderen Systemen zu dokumentieren. Dies geht über die Anforderungen der IEC 62304 für Software-Systeme der Sicherheitsklasse A hinaus. Tipp: Pauschal kann man also nicht sagen, in welchem Umfang Ihre technische Dokumentation erweitert werden muss. Gehen Sie den Anhang IV des AI Acts also am besten durch und prüfen Sie für jeden Punkt, inwiefern Sie diesen durch Ihre aktuelle Dokumentation bereits umgesetzt haben. Alle noch fehlenden Informationen erstellen Sie im Anschluss idealerweise im Zuge der neu definierten bzw. angepassten Prozesse.
4.3 Anforderungen an das Produkt
4.3.1 Protokollierung und Logs
Hochrisiko-KI-Systeme sind zur Protokollierung verpflichtet. Es handelt sich hier also um eine klare technische Vorgabe. Diese Protokollierung muss, wie so oft im “der Zweckbestimmung angemessenen Maße” stattfinden. Was genau das heißt, wird in Artikel 12 der KI-Verordnung definiert. Demnach geht es vor allem um die Aufzeichnung von Ereignissen, die für folgendes relevant sind: a) die Ermittlung von Situationen, die dazu führen können, dass das Hochrisiko-KI-System ein Risiko im Sinne des Artikels 79 Absatz 1 birgt oder dass es zu einer wesentlichen Änderung kommt, b) die Erleichterung der Beobachtung nach dem Inverkehrbringen gemäß Artikel 72 und c) die Überwachung des Betriebs der Hochrisiko-KI-Systeme gemäß Artikel 26 Absatz 5. Im Kern geht es also darum, Situationen, die mit Risiken und Schäden verbunden sind möglichst gut zu überwachen und rückverfolgen zu können. Bei der Umsetzung dieser Protokollierung müssen sich Hersteller an anerkannte Standards und Spezifikationen halten (z.B. Anforderungen des BSI).
4.3.2 Transparenz und Bereitstellung von Informationen für die Nutzer
Wenn Sie noch keine Gebrauchsanweisungen für Ihr Medizinprodukt haben, brauchen Sie spätestens jetzt eine. Sollten Sie bereits eine Gebrauchsanweisung haben, müssen Sie diese vermutlich überarbeiten. Auch wenn es einige Anforderungen gibt, die auch schon von der MDR gestellt werden, fordert der AI Act nun auch Informationen zu z.B. Cyber Security und Risiken für die Grundrechte. Hier müssen auch Genauigkeitsgrade und relevante Genauigkeitskennzahlen des Systems angegeben werden. Prüfen Sie also für Ihre aktuelle Gebrauchsanweisung, inwiefern Sie die Pflichten des AI Acts bereits erfüllen und machen Sie anschließend die notwendigen Anpassungen.
4.3.3 Menschliche Aufsicht
Die Pflicht zur menschlichen Aufsicht ist eine verpflichtende Risikokontrollmaßnahme für Hochrisiko-KI-Systeme. Das System muss so konzipiert und entwickelt werden, dass es von einem Menschen wirksam beaufsichtigt werden kann. Hier lassen sich einige Parallelen zu den Transparenzanforderungen erkennen. Im Grunde muss es einem Nutzer möglich sein, die Vorgänge im Produkt, sowie dessen Ausgaben zu verstehen. Zudem muss er in der Lage sein, das System abzuschalten oder in den Betrieb einzugreifen. Die Möglichkeit zur Aufsicht kann dabei entweder direkt in die Software eingebaut werden, oder auch an den Nutzer abgegeben werden. Grob zusammengefasst muss es schlichtweg möglich sein, Anomalien oder Fehlverhalten zu erkennen und das System im Zweifelsfall stoppen zu können.
4.3.4 Genauigkeit, Robustheit und Cybersicherheit
Hochrisiko-KI-Systeme müssen widerstandsfähig gegenüber Fehlern, Störungen oder Unstimmigkeiten sein, die innerhalb des Systems oder der Umgebung, in der das System betrieben wird, insbesondere wegen seiner Interaktion mit natürlichen Personen oder anderen Systemen auftreten können. Die Robustheit von Hochrisiko-KI-Systemen kann durch technische Redundanz erreicht werden, was auch Sicherungs- oder Störungssicherheitspläne umfassen kann. Hochrisiko-KI-Systeme, die nach dem Inverkehrbringen (oder der Inbetriebnahme) kontinuierlich weiterlernen, sind so zu entwickeln, dass auf möglicherweise verzerrte Ergebnisse mit geeigneten Risikominderungsmaßnahmen eingegangen wird. Hochrisiko-KI-Systeme müssen widerstandsfähig gegen Versuche unbefugter Dritter sein, ihre Verwendung oder Leistung durch Ausnutzung von Systemschwachstellen zu verändern. Die technischen Lösungen zur Gewährleistung der Cybersicherheit von Hochrisiko-KI-Systemen müssen den jeweiligen Umständen und Risiken angemessen sein. Die technischen Lösungen für den Umgang mit KI-spezifischen Schwachstellen umfassen gegebenenfalls Maßnahmen zur Verhütung und Kontrolle von Angriffen, mit denen versucht wird, den Trainingsdatensatz zu manipulieren („Datenvergiftung“), von Eingabedaten, die das Modell zu Fehlern verleiten sollen („feindliche Beispiele“), oder von Modellmängeln.
4.3.5 CE-Kennzeichnung
Eine CE-Kennzeichnung hat Ihr Medizinprodukt bereits, wenn Sie es konform im Rahmen der MDR zugelassen haben. Sie müssen nur noch die entsprechende Verordnung referenzieren, damit Nutzer nachvollziehen können, dass Ihr Produkt im Einklang mit den beiden Verordnungen (MDR und AI Act) entwickelt worden ist.
4.3.6 Registrierung des Produkts
Ähnlich wie EUDAMED für Medizinprodukte, wird es in Zukunft auch eine EU-Datenbank für Hochrisiko-KI-Systeme geben, in welcher Ihr Produkt registriert werden muss. Zum aktuellen Zeitpunkt ist eine solche aber noch nicht öffentlich zugänglich (Stand 20.01.2026).
5. Wie beeinflusst der AI Act die Zulassung meines Medizinprodukts?
Die Risikoklasse Ihres Medizinprodukts nach MDR hat Einfluss auf die Anwendbarkeit des AI Acts. Wie Sie die Risikoklasse nach MDR bestimmen, erfahren Sie in unserem Leitfaden zur Klassifizierung von Software-Medizinprodukten.
5.1 Risikoklasse I
Bei der Zulassung von Produkten der Risikoklasse I nach MDR ändert sich durch den AI Act nichts (bis auf die Anforderungen aus Kapitel 3.3) – solange das Produkt kein Hochrisiko-KI-System darstellt. Sollte dies aber der Fall sein, brauchen Sie für die Konformitätsbewertung in Zukunft eine benannte Stelle, welche die Konformität mit der KI-Verordnung prüft. Behalten Sie dies unbedingt im Hinterkopf, wenn Sie die Umsetzung eines Produkts der Risikoklasse I planen, da es den Markteintritt maßgeblich verzögern kann.
Achtung: Es gelten Ausnahmen für Produkte der Risikoklasse Im, Is und Ir, bei denen eine benannte Stelle in die Konformitätsbewertung involviert ist. Diese sind bei reinen Software-Produkten allerdings eher selten.
5.2 Risikoklasse IIa oder höher
Produkte der Risikoklasse IIa oder höher sind im Grunde automatisch auch Hochrisiko-KI-Systeme gemäß dem AI Act. Somit sind einige Zusatzanforderungen umzusetzen, die weiter oben im Artikel bereits ausgeführt wurden. Bei der Zulassung (insbesondere der Konformitätsbewertung) müssen Sie in Zukunft unbedingt darauf achten, dass Ihre benannte Stelle auch unter der KI-Verordnung akkreditiert ist, oder eine solche Akkreditierung anstrebt. Diese muss im Zuge der Konformitätsbewertung dann nicht nur die Konformität mit der MDR, sondern auch mit dem AI Act prüfen.
Die Konformitätsbewertung wird dann voraussichtlich aber nicht für beide Verordnungen separat, sondern im Zuge einer integrierten Prüfung durchgeführt.
Nur, wenn das Produkt mit beiden Verordnungen konform ist, darf es das CE-Symbol tragen. Setzen Sie sich also schnellstmöglich mit Ihrer benannten Stelle in Verbindung und klären Sie, ob eine solche Akkreditierung für die KI-Verordnung geplant ist.
Die MDCG hat im Juni 2025 in diesem Zusammenhang ein Dokument veröffentlich, welches das Zusammenspiel zwischen MDR und AI Act beschreibt: MDCG 2025-6
6. Ab wann gilt der AI Act? Umsetzungs-Timeline
Der EU AI Act tritt nicht sofort komplett in Kraft, sondern erst über die Zeit. Die aktuelle Timeline sieht vor, dass der Großteil der Verordnung bis 2. August 2026 in Kraft getreten ist und sie ab dem 2. August 2027 dann vollständig gültig ist.
Das geht aus der Timeline der Europäischen Kommission hervor:

Umsetzungs-Zeitplan für den AI Act der Europäischen Kommission (Quelle: AI Act implementation timeline)
Zusammengefasst bedeutet dies für Hersteller von KI- bzw. Software-Medizinprodukten (MDAI):
- Viele grundsätzliche Regeln des AI Act sind bereits heute gültig (z.B. Verbotene KI-Praktiken)
- Ab August 2026 gilt fast der komplette AI Act, allerdings mit einer wichtigen Ausnahme: Artikel 6(1). Dieser beschreibt die Einstufungsregeln für Hochrisiko-KI-Systeme.
- Hersteller von Hochrisiko-KI-Systemen haben somit bis August 2027 Zeit, alle Anforderungen an ihr Produkt und an sich selbst als Hersteller umzusetzen (außer, es ist ein System, welches in Anhang III genannt ist). Denn erst ab diesem Zeitpunkt gilt die Einstufung als Hochrisiko-KI-System.
7. Fazit
Der AI Act unterscheidet sich von der MDR zu allererst durch seine Zielsetzung. Während die MDR v.a. darauf abzielt, die Sicherheit und Funktionstauglichkeit von Medizinprodukten sicherzustellen, schließt die KI-Verordnung auch den Schutz der Grundrechte von Personen mit ein. Grundsätzlich lässt sich sagen, dass vor allem auf MDR-Medizinprodukte der Risikoklasse IIa oder höher viele neue Anforderungen zukommen. Diese werden nämlich automatisch der Kategorie “Hochrisiko-KI-Systeme” gemäß des AI Acts zugeordnet. Doch auch Medizinprodukte der Risikoklasse I können im Einzelfall in diese Kategorie fallen. Für Produkte, die keine Hochrisiko-KI-Systeme darstellen und nicht zu den “verbotenen Praktiken” zählen, bringt die KI-Verordnung wenig Änderungen. Hersteller von Hochrisiko-KI-Medizinprodukten müssen einer Reihe von Stellen Anpassungen vornehmen, um die Anforderungen des AI Act einzuhalten. Neben den Anforderungen des AI Acts, müssen Sie natürlich auch die Anforderungen der MDR beachten, welche in Bezug auf künstliche Intelligenz relevant sind.
Benötigen Sie Unterstützung bei der Entwicklung eines medizinischen KI-Systems? Wir entwickeln medizinische KI-Systeme für Unternehmen im Gesundheitswesen auf Auftragsbasis.
Unsere KI-Services: – Technischer Review von medizinischen KI-Systemen – Entwicklung von neuen Machine Learning-Modellen im Gesundheitsbereich – Individuelle Beratung bezüglich regulatorischer Compliance für Ihr geplantes KI-basiertes Produkt
Wir helfen Ihnen, auch die umfangreichen Anforderungen der MDR und des AI Act umzusetzen und ein erfolgreiches Produkt zu entwickeln.
The post AI Act: Leitfaden für Medizinprodukt-Hersteller nach MDR (2026) appeared first on QuickBird Medical.
]]>The post Interoperabilität für digitale Gesundheitsanwendungen (DiGA) appeared first on QuickBird Medical.
]]>“Μια DiGA πρέπει να πληροί ορισμένες απαιτήσεις διαλειτουργικότητας.”
Verstehen Sie diesen Satz? Sofern Sie kein Griechisch sprechen, vermutlich nicht. Daher benötigen Sie eine deutsche Übersetzung. Das wäre Ihre Interoperabilitätsanforderung an diesen Blogartikel. Wenn wir diese Anforderung umsetzen, lautet der Satz wie folgt:
“Eine DiGA muss bestimmten Interoperabilitätsanforderungen genügen.”
Genauso wie dieser Blogartikel muss auch eine DiGA mit angrenzenden Systemen kompatibel sein und ihr Datenaustausch muss konkreten Regeln folgen: der strukturellen, syntaktischen, semantischen und organisatorischen Interoperabilität. Was es mit diesen Begriffen auf sich hat, erfahren Sie hier. Lesen Sie weiter und erhalten Sie einen praxisnahen Einblick in die Umsetzung von Interoperabilitätsanforderungen an eine DiGA.
Übersicht
- 1. Schnittstellen von DiGA zu anderen Systemen
- 2. Anbindung der elektronischen Patientenakte (ePA)
- 3. Digitale Identität/GesundheitsID
- 4. Fazit
1. Schnittstellen von DiGA zu anderen Systemen
Je nach Einsatzzweck und Funktionsumfang einer DiGA kann diese Schnittstellen zu zahlreichen anderen Systemen aufweisen. Zum Beispiel könnte eine DiGA Daten aus Google Fit oder Apple Health einlesen, Daten zu einem Krankheitsbild an die elektronische Patientenakte (ePA) übergeben, oder eine behandelnde Ärztin zum Therapieverlauf eines Patienten informieren. Nicht jede Schnittstelle muss dabei interoperabel ausgestaltet werden (z.B. zu anderen Apps). Gesetzliche Vorgaben gibt es nur an drei Schnittstellen. Diese werden im folgenden Kapitel näher beschrieben.
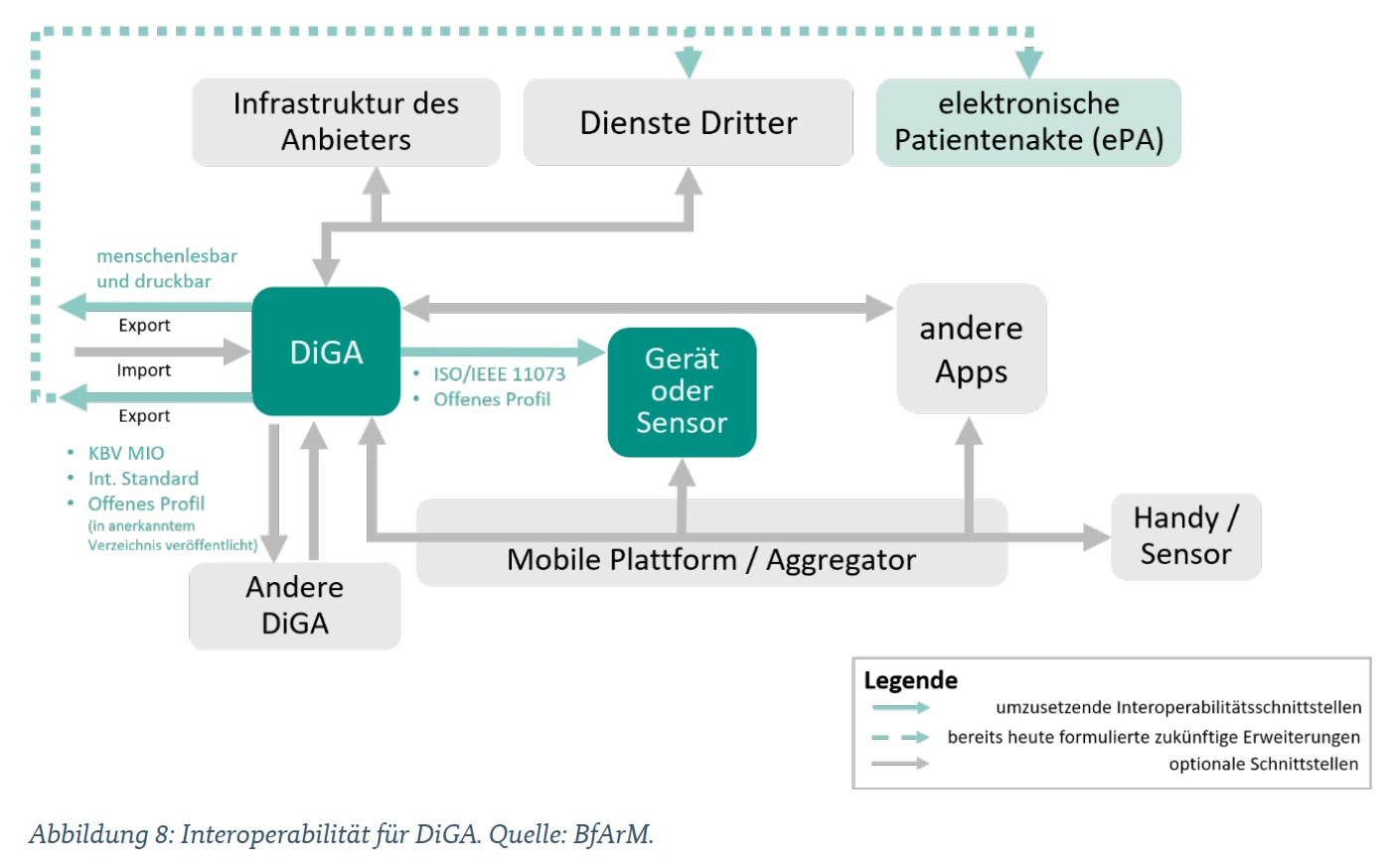
Interoperabilität für DiGA
(Auszug aus dem DiGA Leitfaden – Version 3.6)
1.1 Welche Schnittstellen müssen interoperabel ausgestaltet werden?
Wenn man sich die Kernaufgabe einer DiGA vor Augen führt, die Versorgungsqualität für Patienten zu optimieren, lässt sich schnell begreifen, an welchen Schnittstellen Interoperabilität dabei besonders wichtig ist. Die DiGAV besagt genau, welche Schnittstellen interoperabel ausgestaltet werden müssen. Insbesondere die Anlage 2 zur DiGAV ist für die Umsetzung der Anforderungen von großer Bedeutung.
Notwendige Interfaces für DiGA
Zur Sicherstellung der Versorgung sind in erster Linie Ärzten, Psychotherapeuten und die Patienten selbst von großer Bedeutung.
1. Ein Datenexport in menschenlesbarem, ausdruckbarem Format muss also möglich gemacht werden.
Doch nicht nur Menschen, sondern auch andere Systeme des Gesundheitswesens, wie z.B. die ePA müssen die Daten der DiGA sinnvoll verwenden können.
2. Auch ein Datenexport in einem interoperablen Format muss ermöglicht werden. Die Übermittlung von Daten erfolgt gemäß einer Festlegung für die semantische und syntaktische Interoperabilität. Von Bedeutung ist dabei insbesondere das MIO DiGA-Toolkit der KBV, um Daten in die elektronische Patientenakte (ePA) übertragen zu können.
DiGA, die zum Beispiel auf die Daten von Sensoren und anderen Messgeräten angewiesen sind, müssen auch hier eine interoperable Schnittstelle schaffen, um Hardware-Herstellern eine Anbindung an die DiGA zu ermöglichen.
3. Daher ist auch eine interoperable Schnittstelle zu Wearables und anderen Medizingeräten verpflichtend, die von verschiedenen Herstellern verwendet werden können, um Daten bereitzustellen.
1.1.1 Datenexport in menschenlesbarem, ausdruckbarem Format
Patienten und ihre medizinischen Versorger müssen in der Lage sein, die versorgungsrelevanten Daten einer DiGA zu verstehen und weiterzuverwenden. Aus diesem Grund fordert die DiGAV an dieser Schnittstelle auch eine Möglichkeit, Daten in einem menschenlesbaren, ausdruckbaren Format zur Verfügung zu stellen. Dabei ist es wichtig, dass der Datenexport zwar über die DiGA selbst eingeleitet werden kann, nicht aber durch diese erfolgen muss.
Ein Button, der den Download eines verschlüsselten Datenpakets initiiert, welches später über einen Server abgerufen werden kann, wird hierbei auch als legitime Umsetzung angesehen. Ein Beispiel für einen Datenexport in menschenlesbarem, ausdruckbarem Format wäre die Erstellung einer PDF-Datei, welche alle Blutzuckermesswerte aus der DiGA und andere relevante Daten abbildet.
Der Referentenentwurf zur zweiten Änderung der DiGAV vom 28.10.2025 sieht für die Zukunft außerdem eine Übertragung von diesem menschenlesbaren Export in die elektronische Patientenakte vor.
QuickBird Medical Tipp: Stellen Sie die relevanten Daten in kompakter und verständlicher Form zur Verfügung. Überlegen Sie sich, welche Daten in einem Versorgungsszenario sinnvoll sind, in dem die DiGA zum Einsatz kommt. Welche Daten sind für Patienten und Ärzten von Bedeutung und wie können diese verständlich dargestellt werden?
Abseits der gesetzlichen Interoperabilitätsanforderungen an dieser Schnittstelle, können Sie sich hier einen maßgeblichen Wettbewerbsvorteil verschaffen. Wenn sich Ihre DiGA nämlich gut in den Alltag von Ärzten und Psychotherapeuten integrieren lässt und einen spürbaren Mehrwert liefert, wird diese auch häufiger verschrieben. Je kompatibler Ihre DiGA mit bestehenden Prozessen im Gesundheitswesen ist, desto besser.
1.1.2 Datenexport in interoperablem Format (ePA)
Nicht nur Menschen, sondern auch andere Systeme sind Teil des Gesundheitswesens und wollen die Daten Ihrer DiGA weiterverwenden. Allen voran ist hier die elektronische Patientenakte (ePA) zu nennen, welche Daten von der DiGA in einem interoperablen Format beziehen können muss. Um den Anforderungen der Interoperabilität im Gesundheitswesen gerecht zu werden, ist es sinnvoll, sich auf einheitliche Standards zu einigen, welche zum Datenaustausch verwendet werden. Diese Standards werden von Standardisierungsorganisationen definiert und legen Format und Semantik von Datenströmen fest. Eine Adaption eines Standards für ein bestimmtes Land oder Einsatzfeld nennt man Profil.
Aus genau diesem Grund gibt es konkrete Vorgaben, welche Formate zum Datenexport zulässig sind. Es reicht nicht einfach, dass Ihre DiGA bestimmte Daten zur Verfügung stellt, auch deren Format ist entscheidend. Als DiGA-Hersteller müssen Sie sich nun also auf die Suche nach einem geeigneten Datenformat machen, doch wo sollen Sie anfangen zu suchen?
- Suchen Sie zuallererst für jedes Datenelement nach einem von der KBV definierten medizinischen Informationsobjekt (MIO) aus dem DiGA Toolkit. Dieses sollte in der Regel Ihren Anwendungsfall halbwegs passend abdecken können. Hierfür gibt es auch generischere MIO-Profile wie „Observation_Free“.
- Falls es aber wirklich kein einigermaßen passendes MIO gibt, haben Sie noch zwei Möglichkeiten:
-
- Sie können sich eines anderen existierenden offenen, international anerkannten Schnittstellen- oder Semantikstandards bedienen (z.B. HL7 FHIR).
- Sie können ein eigenes Profil über einen oder mehreren existierenden offenen, international anerkannten Schnittstellen oder Semantikstandard definieren. Dieses Profil muss dann in einem anerkannten Verzeichnis (z.B. FHIR Registry) zur freien Nutzung veröffentlicht werden.
Medizinische Informationsobjekte (MIO) – DiGA Toolkit
Ein MIO ist ein von der KBV definiertes Datenformat für medizinische Informationen. Mit dem großen Ziel, einen optimalen Datenaustausch zwischen allen in der medizinischen Versorgung beteiligten Systeme zu gewährleisten, werden von Zeit zu Zeit neue MIOs für verschiedene Informationen entwickelt. Insbesondere die Schnittstelle zur ePA ist einer der Hauptgründe für die Einführung von MIOs.
Dass Hersteller einen veröffentlichten Interoperabilitäts-Standard verwenden, ist gut. Immerhin wird die Form des Datenaustauschs somit transparent. Noch besser ist es natürlich, wenn der gewählte Standard auch noch zum Standard der ePA oder anderen Systemen passt. Somit ist eine direkte Anbindung möglich.
Ein Beispiel für ein MIO ist der elektronische Impfpass. Seine Struktur ist genau festgelegt und gibt vor, dass neue Impfeinträge einem klaren Format entsprechen müssen, um diese in den Impfpass zu integrieren. Das MIO definiert, welche Informationen enthalten und in welchem Format diese vorliegen müssen.
Speziell für DiGA ist das DiGA Toolkit interessant, welches dafür entwickelt wurde, die Interoperabilitätsanforderungen an DiGA umzusetzen.
Welche Daten müssen für den interoperablen Datenexport berücksichtigt werden?
Theoretisch können unzählige verschiedene Daten aus einer App exportiert werden. Für den interoperablen Datenexport müssen allerdings nur jene Daten berücksichtigt werden, die auf dem bestimmungsgemäßen Gebrauch durch die Nutzer basieren (siehe § 4 Absatz 2 Satz 1 Ziffer 1 DiGAV). Daten des bestimmungsmäßigen Gebrauchs sind solche, die für Zwecke benötigt werden, die in unmittelbarem Zusammenhang mit der Erreichung des medizinischen Nutzens oder von patientenrelevanten Struktur- und Verfahrensverbesserungen stehen.
Konkreter betrifft dies potenziell folgende Daten:
- durch die Nutzer eingegebene Daten
- über Geräte und Sensoren erfasste Daten
- Daten zum Nutzer und zum Nutzungskontext (sofern verfügbar)
- Angaben zur DiGA und Metadaten zum Datenexport
Folgende Daten müssen nicht als eigenständige Objekte exportierbar sein:
- Ableitungen auf erfassten Daten, die ausschließlich zur Sicherstellung des sicheren Betriebs der DiGA dienen.
- Auf den Daten angelegte statistische Verfahren, welche nur zum Nachweis des positiven Versorgungseffekts gespeichert werden.
1.1.3 Schnittstelle zu Wearables & medizinischen Geräten
Diese Schnittstelle ist nicht bei allen DiGA vorhanden und somit nicht für alle Hersteller relevant. Wenn aber Medizingeräte oder andere Wearables an der Funktion der DiGA beteiligt sind, gilt es auch an dieser Schnittstelle, einen interoperablen Datenaustausch zu gewährleisten. Der Grund dafür ist ganz einfach: es muss dem Patienten/der Patientin selbst überlassen werden, welches Gerät er oder sie verwenden möchte. Dazu ist es notwendig, dass Hardware-Herstellern klar ist, in welchem Datenformat mit der DiGA kommuniziert wird. Nur so können Sie Geräte entwickeln, die auch mit der DiGA interagieren können.

Anforderungen an Hardwarehersteller
Zur Umsetzung der Interoperabilität an dieser Schnittstelle haben DiGA-Hersteller folgende Möglichkeiten:
- Nutzung eines offengelegten, dokumentierten Profils des ISO/IEEE 11073-Standards (Medical Device Communication)
- Suchen Sie ein durch die BluetoothSIG spezifiziertes Health Device Profil.
- Nutzung eines offenen, international anerkannten Standards (z. B. HL7 FHIR) oder entsprechender Profile
- Definition eines eigenen Profils auf Basis offener Standards. Dieses muss zur freien Nutzung in einem anerkannten Verzeichnis (z. B. FHIR Registry) veröffentlicht werden. Als nationale Referenz- und Orientierungshilfe dient der INA – Interoperabilitäts-Navigator der gematik.
QuickBird Medical Hinweis: Ihre DiGA darf zusätzlich zu einer interoperablen Schnittstelle auch nicht-interoperable Schnittstellen anbieten. Das kann zum Beispiel der Fall sein, wenn Sie ein Wearable einbinden müssen, welches keine Datenübertragung in einem entsprechenden Format anbietet. Sie dürfen die Schnittstelle zu diesem Gerät aufrechterhalten, müssen aber zudem auch eine interoperable Schnittstelle für andere Geräte schaffen.
Muss eine DiGA auch eine interoperable Schnittstelle zu Geräten anbieten, deren Daten über Apple Health oder Google Fit eingelesen werden? Nein, in diesem Fall muss Ihre DiGA keine interoperable Schnittstelle zum Gerät selbst anbieten.
Auch Hersteller von Hilfsmitteln und Implantaten werden in Zukunft in puncto Interoperabilität in die Pflicht genommen. Hilfsmittel und Implantate, welche Patientendaten an ein Backend des Herstellers oder eine Drittpartei senden, müssen ab 1. Juli 2027 eine interoperable Schnittstelle für DiGA anbieten. So soll es möglich werden, dass DiGA auf Daten aus verschiedenen Quellen zugreifen können. Mehr dazu lesen Sie hier: Guide zum § 374a SGB V – Interoperabilität für DiGA, Hilfsmittel & Implantate
1.2 An welchen Schnittstellen ist die interoperable Ausgestaltung optional?
DiGA können, müssen aber nicht zu allen angrenzenden Systemen eine interoperable Schnittstelle anbieten. Zum Beispiel kann eine DiGA Daten aus anderen Apps, wie Apple Health beziehen, ohne dazu eine interoperable Schnittstelle zu haben. Auch ein direkter Datenaustausch zwischen DiGA und anderen Medical Apps erfordert nicht zwangsläufig eine interoperable Schnittstelle. Sehen Sie sich dazu die Abbildung unter dem Kapitel „Schnittstellen von DiGA zu anderen Systemen“ an. Dort sehen Sie, welche Schnittstellen Ihrer DiGA nicht interoperabel ausgestaltet werden müssen.
2. Anbindung der elektronischen Patientenakte (ePA)
Die Anbindung der DiGA an die elektronische Patientenakte ist ein zentraler Bestandteil der Interoperabilitätsanforderungen. Ziel ist es, versorgungsrelevante Daten aus der DiGA strukturiert, standardisiert und für andere Systeme des Gesundheitswesens nutzbar zu machen.
DiGA müssen den Export von Daten in die ePA in einem interoperablen Format ermöglichen. Dabei sind sowohl eine manuelle Datenübermittlung durch die Versicherten als auch eine regelmäßige automatisierte Übertragung vorzusehen. Die automatisierte Übermittlung muss jederzeit deaktivierbar sein, sodass den NutzerInnen stets beide Optionen zur Verfügung stehen.
Die technische Umsetzung erfolgt auf Basis der von der Kassenärztlichen Bundesvereinigung definierten Medizinischen Informationsobjekte (MIO), insbesondere des DiGA Toolkits, welches die semantischen und syntaktischen Anforderungen für den Datenaustausch mit der ePA festlegt.
Der Nachweis der erfolgreichen ePA-Anbindung in der Referenzumgebung erfolgt im Rahmen eines Bestätigungsverfahrens der gematik und ist Voraussetzung für die formale Vollständigkeit eines Antrags auf Aufnahme in das DiGA-Verzeichnis beim BfArM. Das BfArM testet die Anbindung außerdem im Rahmen der inhaltlichen Prüfung des DiGA-Antrags.
Wir unterstützen Sie gerne bei der technischen Anbindung Ihrer DiGA an die ePA. Mehr Informationen zu diesem Service finden Sie hier.
3. Digitale Identität bzw. GesundheitsID
Um in die ePA zu schreiben und aus dieser zu lesen, muss eine DiGA außerdem die Authentisierung eines Users mit einer digitalen Identität (GesundheitsID) implementieren. Auch wenn diese Anforderung nicht formell unter das Thema „Interoperabilität“ fällt, stellt sie dennoch eine weitere erforderliche Schnittstelle zu anderen Systemen im Gesundheitswesen dar.
Über die GesundheitsID kann eine DiGA die Krankenversichertennummer des Patienten anfragen. Diese ist Voraussetzung für den Export in die ePA. Daher ist eine Übertragung von Daten in die ePA nur möglich, wenn der Patient seinen DiGA-Account mit der GesundheitsID verbunden hat. Außerdem muss der Patient innerhalb seiner ePA-App den Zugriff für die entsprechende DiGA explizit freigeben.
Wir unterstützen Sie gerne bei der Implementierung der GesundheitsID-Schnittstelle. Mehr Informationen zu diesem Service finden Sie hier.
4. Fazit
Interoperabilität in der DiGA-Entwicklung ist auf den ersten Blick ein undurchsichtiges Unterfangen. Wenn man sich genauer damit befasst, lassen sich aber drei zentrale Punkte ableiten:
- Ein Mensch muss die Daten einer DiGA verstehen können (menschenlesbar)
- Andere Systeme (insb. die ePA) müssen Daten einer DiGA verstehen können
- Hersteller von Sensoren und Messgeräten müssen wissen, in welchem Format der Datenaustausch mit der DiGA stattfindet
Bei der Suche nach geeigneten interoperablen Dateiformaten sind die ersten Anlaufstellen für Sie als Hersteller die MIOs der KBV und vor allem das DiGA Toolkit.
Benötigen Sie Hilfe bei der ePA-Anbindung oder GesundheitsID?
Als spezialisierter Dienstleister waren wir bereits in die Umsetzung von über 15 verschiedenen DiGA-Projekten involviert. Wir unterstützen Sie gerne bei der Implementierung aller Interoperabilitäts-Anforderungen (z.B. Anbindung an die ePA). Mehr Informationen zu unserem ePA & GesundheitsID-Service finden Sie hier.
Bei allen weiteren Fragen, kontaktieren Sie uns gern über unser Kontaktformular: Zum Kontaktformular
The post Interoperabilität für digitale Gesundheitsanwendungen (DiGA) appeared first on QuickBird Medical.
]]>The post DiGA-Preisverhandlung mit dem GKV-Spitzenverband appeared first on QuickBird Medical.
]]>Die Informationen aus diesem Artikel stammen u.a. aus dem seit 28.02.2024 gültigen Rahmenbedingungen des GKV-Spitzenverbands.
Stand Januar 2026: Mit dem Digital-Gesetz (DigiG) wurde am 26.03.2024 abschließend festgelegt, dass der Anteil erfolgsabhängiger Preisbestandteile ab dem 01.01.2026 mindestens 20 % des Vergütungsbetrags betragen muss. Auch diese Änderung wird in Zukunft eine wichtige Rolle in Preisverhandlungen spielen. Wir gehen in Kapitel 4 im Detail auf diese Neuerung ein.
Inhaltsverzeichnis
- 1. Die Timeline der DiGA-Preisverhandlung
- 2. Wann gilt welcher Preis für die DiGA?
- 3. Die Preisverhandlung mit dem GKV-Spitzenverband
- 4. Anwendungsbegleitende Erfolgsmessung (AbEM) für DiGA
- 5. Weitere Fragen?
1. Die Timeline der DiGA-Preisverhandlung
Es macht einen Unterschied, ob Sie eine dauerhafte oder vorläufige Aufnahme für Ihre DiGA beantragen. Je nach Antrag beginnen die Verhandlungen früher oder später.
Wenn Ihre DiGA gleich dauerhaft aufgenommen wird (ohne Erprobungsjahr):
- Der Verhandlungszeitraum beginnt, nachdem ihre DiGA 4 Monate im Verzeichnis gelistet ist.
- Sie übermitteln dem GKV-Spitzenverband alle notwendigen Unterlagen.
- Nach 5 weiteren Monaten ist die DiGA-Preisverhandlung abgeschlossen.
- Falls es zu keiner Einigung kommt, entscheidet in weiteren 3 Monaten die Schiedsstelle über den Vergütungsbetrag.
- Der verhandelte oder durch die Schiedsstelle festgelegte Vergütungsbetrag ist grundsätzlich ab dem 13. Monat nach Eintragung der DiGA im Verzeichnis als Vergütung anzusetzen. Bis dahin gilt der tatsächliche Herstellerpreis. Wird der Vergütungsbetrag erst später festgelegt, erfolgt eine rückwirkende Korrektur bzw. Rückzahlung ab dem 13. Monat.
Wenn Ihre DiGA zuerst vorläufig aufgenommen wird (mit Erprobungsjahr):
- Spätestens zum Ende der Erprobungsphase (in der Regel 12 Monate; ggf. verlängert) übermitteln Sie dem BfArM die erforderlichen Nachweise zum positiven Versorgungseffekt.
- Nach spätestens weiteren 3 Monaten erhalten Sie einen Bescheid des BfArM über die dauerhafte Aufnahme und der Verhandlungszeitraum beginnt.
- Sie übermitteln dem GKV-Spitzenverband alle notwendigen Unterlagen und einigen sich auf Verhandlungstermine.
- Nach weiteren 5 Monaten ist die DiGA-Preisverhandlung abgeschlossen. Falls es zu keiner Einigung kommt, entscheidet in weiteren 3 Monaten die Schiedsstelle über den Vergütungsbetrag.
- Der verhandelte oder durch die Schiedsstelle festgelegte Vergütungsbetrag wird grundsätzlich ab dem 13. Monat nach (vorläufiger) Eintragung der DiGA im Verzeichnis vergütet. Bis zur Festlegung gilt der tatsächliche Herstellerpreis. Wird der Vergütungsbetrag erst später festgelegt, erfolgt eine rückwirkende Korrektur ab dem 13. Monat mit entsprechenden Ausgleichszahlungen.
2. Wann gilt welcher Preis für die DiGA?
Der tatsächliche Herstellerpreis gilt ab dem Tag der Eintragung ins DiGA-Verzeichnis bis zur geschlossenen Vergütungsvereinbarung.
- Er gilt 12 Monate (unabhängig davon, ob die DiGA zunächst vorläufig oder sofort dauerhaft aufgenommen wurde).
- Bereits im ersten Jahr können Höchstbeträge zu beachten sein.
- Der Hersteller kann den tatsächlichen Preis innerhalb dieser 12 Monate einmal ändern.
Ab dem 13. Monat wird grundsätzlich der verhandelte oder durch die Schiedsstelle festgelegte Vergütungsbetrag vergütet. Wird der Vergütungsbetrag erst später vereinbart oder festgesetzt, wirkt er rückwirkend ab dem 13. Monat. Daraus können Ausgleichszahlungen resultieren.
3. Die Preisverhandlung mit dem GKV-Spitzenverband
3.1 Beginn der Verhandlungen
Der Start der DiGA-Preisverhandlung mit dem GKV-Spitzenverband hängt davon ab, ob Ihre DiGA mit oder ohne Erprobungsjahr ins Verzeichnis aufgenommen wurde. Aus diesem Grund gibt es zwei Fälle:
3.1.1 Ihre DiGA ist von Anfang an dauerhaft gelistet
In diesem Fall beginnt die Preisverhandlung 4 Monate nach der Aufnahme ins DiGA-Verzeichnis.
Hinweis: Durch das Inkrafttreten des DVPMG am 09.06.2021 wurde der Beginn der Verhandlungen von 6 auf 4 Monate verkürzt.
3.1.2 Ihre DiGA ist vorläufig gelistet
Wenn Ihre DiGA erst vorläufig ins Verzeichnis aufgenommen wurde, beginnen die Preisverhandlungen erst, sobald ein Bescheid über die endgültige Aufnahme vorliegt. Bei einem regulären Erprobungsjahr (12 Monate) und einer Bearbeitungszeit von bis zu 3 Monaten liegt dieser Bescheid typischerweise bis zum 15. Monat vor. Bei einer verlängerten Erprobungsphase entsprechend später.
3.2 Notwendige Unterlagen
- Bescheid des BfArM zur Aufnahme ins DiGA-Verzeichnis
- CE-Konformitätskennzeichnung oder die CE-Kennzeichnung
- Erklärung zu allen in der DiGAV Anlage 1 und Anlage 2 genannten Punkten (wenn vorhanden auch entsprechende Zertifikate)
- Studienberichte zum Nachweis der positiven Versorgungseffekte (ggf. auch Publikationen)
- Zahl der bis zu 5 Tage vor der Übermittlung der Unterlagen eingelösten Freischaltcodes/Rezeptcodes
- Informationen zu Preisen für die DiGA bei Abgabe an Selbstzahler
- Informationen zu Preisen für die DiGA in anderen europäischen Ländern, die dort von Kostenträgern übernommen werden
- Bei DiGA, die vorläufig aufgenommen wurden: die festgelegten Nachweise zur Evaluation, welche Sie auch dem BfArM übermittelt haben
Sie können auch noch andere Unterlagen einreichen, die zur Verhandlung eines Vergütungsbetrags dienlich sind. Bis zu 10 Tage vor dem ersten bzw. zweiten Verhandlungstermin können Sie beispielsweise noch folgende Unterlagen nachreichen:
- Anwendungsbegleitende Auswertungen der DiGA
- Studien zu positiven Versorgungseffekten
- Auswertungen von Leistungs- und Abrechnungsdaten
3.3 Dauer der Verhandlungen
Die DiGA-Preisverhandlung mit dem GKV-Spitzenverband dauert 5 Monate. Innerhalb dieser fünf Monate finden 3 Verhandlungstermine im Umfang von maximal 3 Stunden statt. Es besteht die Möglichkeit, sich auf einen zusätzlichen vierten Termin zu einigen.
Hinweis: Durch das Inkrafttreten des DVPMG am 09.06.2021 wurde der Verhandlungszeitraum von 6 auf 5 Monate verkürzt.
Sollte es zu keiner Einigung kommen, wird nach der Verhandlungsphase die Schiedsstelle eingeschaltet, welche alle Unterlagen prüft und nach spätestens 3 Monaten über einen Vergütungsbetrag entscheidet. Die Kosten für das Schiedsstellenverfahren werden zwischen DiGA-Hersteller und GKV-Spitzenverband aufgeteilt.
3.4 Wichtige Faktoren zur Ermittlung des Vergütungsbetrags
Der Vergütungsbetrag ermittelt sich auf Basis aller preisrelevanten Unterlagen und der im Verzeichnis öffentlichen Informationen. Für Sie als Hersteller ist aber wichtig zu wissen, dass die Stärke des Versorgungseffekts eine besondere Rolle für die Höhe des Betrags spielt. Aussagekräftige Daten, Studien und Publikationen dürften Ihnen hier also die meisten Vorteile verschaffen.
Was Sie beim Nachweis positiver Versorgungseffekte beachten müssen, erfahren Sie hier: DiGA: Leitfaden zum Nachweis positiver Versorgungseffekte
Auch die Preise vergleichbarer DiGA und Anwendungen werden einen Einfluss auf Ihren Vergütungsbetrag haben. Erarbeiten Sie also auch Argumente, die den Mehrwert Ihrer DiGA gegenüber anderen Angeboten unterstreichen.
Womöglich limitiert wird der Vergütungsbetrag allerdings durch gruppenspezifische Höchstbeträge.
3.5 Weitere Informationen zur Preisverhandlung
Die Informationen in diesem Artikel sind zugunsten der Leserlichkeit bewusst vereinfacht dargestellt. Für eine detaillierte, rechtssichere Einordnung der DiGA-Preisverhandlung sind insbesondere die folgenden Quellen maßgeblich:
- GKV-Spitzenverband – Rahmenvereinbarung nach § 134 Abs. 4 und 5 SGB V: Zentrale Rechtsgrundlage für Ablauf, Fristen, Verhandlungsmechanik, Vergütungs- und Höchstbeträge sowie Ausgleichsregelungen im Rahmen der DiGA-Preisverhandlung.
- Bundesinstitut für Arzneimittel und Medizinprodukte – DiGA-Leitfaden des BfArM: Erläutert die Anforderungen an die Aufnahme ins DiGA-Verzeichnis, die Erprobungsphase sowie den Nachweis positiver Versorgungseffekte als Grundlage für die dauerhafte Listung.
Diese Dokumente sind für die konkrete Auslegung und Anwendung der Regelungen stets heranzuziehen.
4. Anwendungsbegleitende Erfolgsmessung (AbEM) für DiGA
Mit dem Digital-Gesetz (DigiG) wurde die anwendungsbegleitende Erfolgsmessung (AbEM) als Bestandteil der DiGA-Vergütungssystematik in § 134 SGB V verankert.
Für Vergütungsvereinbarungen, die ab dem 1. Juli 2026 neu abgeschlossen oder durch die Schiedsstelle festgesetzt werden, ist ein erfolgsabhängiger Vergütungsanteil verpflichtend vorgesehen. Dieser muss mindestens 20 % des Vergütungsbetrags umfassen und auf einer anwendungsbegleitenden Erfolgsmessung basieren.
Die AbEM beschreibt die systematische Erhebung und Auswertung von Nutzungs- und Ergebnisdaten einer DiGA im Versorgungsalltag. Ziel ist es, den tatsächlichen Versorgungseffekt unter Realbedingungen nachvollziehbar abzubilden und als Grundlage für die Ausgestaltung und Bewertung erfolgsabhängiger Preisbestandteile heranzuziehen.
Die AbEM ersetzt nicht den Nachweis positiver Versorgungseffekte für die Aufnahme in das DiGA-Verzeichnis. Sie kann diesen jedoch im Rahmen der Preisverhandlung ergänzen und zur weiteren Bewertung von Nutzung, Wirksamkeit und patientenrelevanten Effekten beitragen.
Wie genau DiGA-Hersteller diese Erfolgsmessung implementieren werden und, ob der variable Anteil zu einer durchschnittlichen Erhöhung oder Minderung des Vergütungsbertrags führen wird, ist aktuell noch nicht klar. Wir informieren Sie in unserem DiGA-Newsletter, sobald hierzu Neuigkeiten bekannt werden.
5. Weitere Fragen?
Als Dienstleister für die Entwicklung von DiGA und Medical Software sind wir mit den aktuellen gesetzlichen Bestimmungen für DiGA sehr vertraut. Daher finden Sie in unserem Blog auch zahlreiche weitere Fachartikel zur DiGA-Entwicklung.
Sie planen die Entwicklung einer DiGA? Sprechen Sie uns gerne an: Zum Kontaktformular
The post DiGA-Preisverhandlung mit dem GKV-Spitzenverband appeared first on QuickBird Medical.
]]>The post DiGA Umsatz & Kosten: Lohnt sich eine DiGA? appeared first on QuickBird Medical.
]]>Es stellt sich eine zentrale Frage: Lohnt sich die Entwicklung Ihrer DiGA-Idee für Sie?
Diese Frage stellen sich auch potenzielle Investoren für Ihr DiGA-Vorhaben. Daher sollten Sie eine Kalkulation mit belastbaren Zahlen vorweisen können.
Besonders relevant sind hierbei folgende Kennzahlen:
- Gewinn
- Umsatz (Umsatzpotenzial & Marktpotenzial bzw. TAM, SAM, SOM)
- Kosten (Entwicklung, Studie, Vermarktung etc.)
Mit diesem Artikel geben wir Ihnen eine detaillierte Anleitung an die Hand, um das Potenzial Ihrer DiGA für Ihre angestrebte Krankheitsindikation zu berechnen. Sie finden außerdem eine Vielzahl von Beispielrechnungen, um die Thematik für Sie möglichst greifbar zu machen.
Inhalte dieses Artikels
- 1. Gewinn & Return on Investment (ROI) einer DiGA
- 2. Kosten für die Entwicklung und Vermarktung einer DiGA
- 3. Download: Excel-Planner zur Prognose von Umsatz & Kosten Ihrer DiGA
- 4. Update 2026: Anwendungsbegleitenden Erfolgsmessung (AbEM) bei DiGA
- 5. Fazit
1. Gewinn & Return on Investment (ROI) einer DiGA
Wir fangen mit der Prämisse an, dass Ihr DiGA-Vorhaben das Ziel hat, profitabel zu sein. Um zu entscheiden, ob Ihr DiGA-Case profitabel sein kann, sollten Sie den zukünftigen Gewinn bzw. Return on Investment (ROI) abschätzen.
Auch die Berechnung des Gewinns einer DiGA entzieht sich dabei nicht den betriebswirtschaftlichen Grundprinzipien und folgt der simplen Formel:
Gewinn = Umsatz – Kosten
Der Return on Investment (ROI) wird wie folgt kalkuliert:
ROI = Gewinn/Kosten
Nun stellt sich natürlich die Frage: Wie konkret lassen sich „Umsatz“ und „Kosten“ prognostizieren? Auf diese Frage gehen wir in den folgenden Abschnitten im Detail ein.
1.1 Umsatz & Marktpotenzial
Der Umsatz entsteht, sobald Patienten die DiGA durch Einlösung eines Freischaltcodes nutzen. Mit jeder Einlösung erhalten Sie als Hersteller Geld von den Krankenkassen in Höhe des Preises Ihrer DiGA.
Den Freischaltcode erhalten Patienten von der Krankenkasse. Dies geschieht über zwei Wege:
- Verschreibung durch Arzt: Der Arzt verschreibt die DiGA an den Patienten auf Basis einer gestellten Diagnose
- Direktweg zur Krankenkasse: Der Patient beantragt einen Freischaltcode bei der Krankenkasse auf Basis einer bereits gestellten Diagnose
Wichtig: Sie erhalten nur Geld, wenn der Patient am Ende erfolgreich einen Freischaltcode erhält und auch in Ihrer DiGA einlöst.
Die Formel für den Umsatz einer DiGA ist demnach:
Umsatz = DiGA-Preis * Freischaltcode-Einlösungen
Weitere mögliche Umsatz-Quellen für DiGA (B2B-Kooperationen, Selbstzahlermodelle etc.) behandeln wir der Einfachheit halber nicht in diesem Artikel.
Beginnen wir mit dem DiGA-Preis: wie können Sie prognostizieren, was der DiGA-Preis für Ihr Indikationsgebiet sein wird?
1.2 Preis bzw. Vergütungsbetrag
Nach der vorläufigen Aufnahme einer DiGA gilt innerhalb des ersten Jahres ein vom Hersteller festgelegter Preis (in Ausnahmefällen verlängerbar auf 2 Jahre). Dieser Preis wird nach Ablauf des Erprobungszeitraums mit dauerhafter Aufnahme der DiGA neu verhandelt und erfahrungsgemäß stark verringert. Der anfängliche Hersteller-Preis ist daher für den langfristigen Erfolg einer DiGA irrelevant und im Rahmen dieser Kalkulation vernachlässigbar.
Der finale Preis bzw. der Vergütungsbetrag einer DiGA wird zwischen dem DiGA-Hersteller und dem GKV-Spitzenverband verhandelt.
Der durchschnittliche Vergütungsbetrag aller dauerhaft gelisteten DiGA beträgt etwa 221 Euro (Stand 2024). Wie in der folgenden Grafik sichtbar, weicht auch keine DiGA stark von diesem Durchschnittspreis ab. Der niedrigste Vergütungsbetrag liegt bei 189 € (Kalmeda) und der höchste Betrag bei 243 € (elevida).
Sie können also davon ausgehen, dass auch Ihre DiGA mit hoher Wahrscheinlichkeit mit einem Betrag innerhalb dieser Spanne vergütet werden wird. Die wichtigsten Informationen zur DiGA-Preisverhandlung mit dem GKV-Spitzenverband finden Sie in diesem Blogbeitrag.
Wichtig ist hierbei auch zu erwähnen, dass der Preis Ihrer DiGA bestimmte Höchstbeträge (mit wenigen Ausnahmen) nicht überschreiten darf: Höchstbeträge und Schwellenwerte.

Verhältnis: Vergütungsbeträge dauerhaft gelisteter DiGA vs. ursprüngliche Herstellerpreise
Quelle: Daten des GKV-Spitzenverbandes gem. § 33a Abs. 6 SGB V; Stand 30.09.2023
QuickBird Medical – Empfehlung
Wir empfehlen eher konservativ zu rechnen, um später keine böse Überraschung zu haben, wenn der Best-Case Ihrer Kalkulation doch nicht eintritt. Kalkulieren Sie Ihren Case mal mit 200 Euro, um zu sehen, ob sich Ihr Vorhaben auch mit diesem Preis lohnen würde.
1.3 Anzahl der Freischaltcode-Einlösungen
Um die zukünftige Anzahl an Verschreibungen bzw. Nutzern einer DiGA abschätzen zu können, lohnt es sich, einen genaueren Blick auf die Prävalenz (Anteil der erkrankten Personen an der Gesamtpopulation) einer bestimmten Indikation zu werfen. Wenn wir in diesem Beitrag von „Prävalenz“ sprechen, beziehen wir uns immer auf die „12-Monatsprävalenz“.
Die Prävalenz kann aus epidemiologischen Datenbanken oder Gesundheitsberichten wie dem Robert Koch-Institut oder Statistiken des Bundesgesundheitsministeriums gewonnen werden. Eine Google-Recherche ist hierfür ein guter Startpunkt.
Sobald Sie die Prävalenz(-Rate) bestimmt haben, multiplizieren Sie diese mit der Anzahl Gesetzlichversicherten in Deutschland. Wenn Sie dies mit dem prognostizierten Preis multiplizieren (siehe oben) erhalten Sie einen groben Schätzwert für das Marktpotenzial Ihrer DiGA.
Maximale-Freischaltcode-Einlösungen = Prävalenz-Rate x Anzahl-Gesetzlichversicherter
Vereinfachtes Beispiel für den Adipositas-DiGA-Gesamtmarkt
(exklusive Folgeverordnungen)
2022 lag die Prävalenz von Adipositas in Deutschland bei 19 %. Bei aktuell ca. 74 Millionen Versicherten ergibt sich Folgendes:
Maximale-Freischaltcode-Einlösungen = 0,19 x 74.000.000 = 14.060.000
(= ca. 14 Millionen Code-Einlösungen pro Jahr)
Weiter unten im Artikel gehen wir außerdem noch auf den Einfluss von Folgeverordnungen auf den Gesamtmarkt ein.
1.4 Marktdurchdringungsrate
Bisher haben wir das maximale Umsatzpotenzial einer DiGA berechnet. Es ist jedoch nicht realistisch, dass Sie jeden einzelnen Patienten in Deutschland mit Ihrer DiGA erreichen werden. Im nächsten Schritt sollten Sie daher noch die Marktdurchdringungsrate abschätzen. Das ist der Prozentsatz der Patienten, die sie planen auch realistisch gesehen zu erreichen.
Für die Abschätzung der Marktdurchdringungsrate müssen Sie eine Vielzahl von Faktoren berücksichtigen: haben Sie bereits Zugang zur Patientengruppe oder den Behandlern? Wenn ja, wie viele Personen können Sie so erreichen? Wie technisch affin ist Ihre Zielgruppe und wer benützt auch wirklich ein Smartphone? Wie stark ist die Konkurrenz anderer DiGA-Lösungen? Wie oft werden gelistete DiGA in diesem Bereich aktuell verschrieben? usw.
Da die Marktdurchdringungsrate sehr individuell für jedes Unternehmen berechnet werden muss, können wir Ihnen hier keinen allgemeingültigen Richtwert an die Hand geben.
Beispiel auf Basis der Marktdurchdringungsrate
Wenn Sie eine prognostizierte Marktdurchdringungsrate von 10 % schätzen, kommen Sie bei einem Gesamtmarkt von 14.060.000 möglichen Freischaltcode-Einlösungen (siehe vorheriges Beispiel) bei Adipositas auf:
Prognostizierte Freischaltcode-Einlösungen = 0,1 x 14.060.000= 1.406.000
(ca. 1,4 Millionen Code-Einlösungen pro Jahr)
Das Beispiel veranschaulicht, wie die Kalkulation in der Praxis aussehen könnte.
QuickBird Medical – Empfehlung
Sehen Sie sich die Verschreibungszahlen anderer DiGA in den aktuellen DiGA-Berichten an, um ein Gefühl zu bekommen, wie oft DiGA in der Praxis aktuell wirklich verschrieben werden. In der folgenden Abbildung finden Sie eine Übersicht aus dem Jahre 2023.

Verordnungen für DiGA mit mehr als 5 Tsd. Verordnungen
Quelle: Daten des GKV-Spitzenverbandes gem. § 33a Abs. 6 SGB V – Stand 30.09.2023
1.5 Folgeverordnungen von DiGA
Es ist relevant auch einmal auf den Unterschied zwischen Erstverordnungen und Folgeverordnungen einzugehen, um dessen Bedeutung für die Kalkulation richtig einzuordnen.
Von einer Erstverordnung sprechen wir, wenn die DiGA zum ersten Mal verschrieben bzw. eingelöst wird. Eine Folgeverordnung kommt dann infrage, wenn mit der Erstverordnung das anvisierte Therapieziel nicht erreicht wurde. In diesem Fall können manche DiGA wiederholt verschrieben werden (falls es sich nicht um eine sogenannte Einmallizenz handelt).
Besonders wichtig ist es hierbei zu verstehen, dass 83 Prozent aller eingelösten Verordnungen zu den Erstverordnungen einer DiGA zählen. Demnach ergibt sich ein Großteil des DiGA-Umsatzes aus Erstverordnungen. Bei DiGA mit Einmallizenz ist eine Folgeverordnung sogar gänzlich ausgeschlossen.

Folgeverordnungen je DiGA
Quelle: Daten des GKV-Spitzenverbandes gem. § 33a Abs. 6 SGB V; Stand 30.09.2023
Bei der Prognosse Ihres DiGA-Umsatzes können Sie Folgeverordnungen in die prognostizierten Zahlen auf Basis der Krankheitsprävalenz einkalkulieren.
Vereinfachtes Beispiel für den Adipositas-DiGA-Gesamtmarkt
(inklusive Folgeverordnungen)
2022 lag die Prävalenz von Adipositas in Deutschland bei 19%. Bei ca. 75 Millionen Versicherten (Stand Januar 2026) und eine Folgeverordnungsquote von 34% der DiGA „Zanadio“ ergibt sich:
Maximale-Freischaltcode-Einlösungen = 0,19 * 74.000.000 * 1,34 = 18.840.400
(= ca. 19 Millionen Code-Einlösungen pro Jahr)
1.6 Beispiel: Prognose des DiGA-Umsatzes für Adipositas
Wichtige Aspekte der Umsatzprognose für DiGA haben wir in den vorherigen Kapiteln erklärt. Nun gehen wir die Formeln in einem Beispiel-Case im Adipositas-Bereich mit Ihnen durch.
Wichtig ist hierbei zu betonen, dass dieses Beispiel nur der Verständlichkeit dient. Kennzahlen wie die Marktdurchdringungsrate müssen individuell im Kontext Ihres Unternehmens kalkuliert werden.
Wir gehen von folgenden Kennzahlen für eine Adipositas-DiGA in Deutschland aus:
Anzahl der gesetzlich Versicherten: 74 Millionen
Prävalenz von Adipositas: 19 %
Prognostizierte Folgeverordnungsquote von Adipositas: 34 %
Prognostizierter DiGA-Vergütungsbetrag: 200 €
Beispielhaft geschätzte Marktdurchdringungsrate: 10 %
Somit ergeben sich folgende Kennzahlen für das Unternehmen:
Marktpotenzial bzw. TAM (Total Addressable Market)
= 74 Millionen x 0,19 x 1,34 x 200 € = 3.768.080.000 €
(3,7 Milliarden Euro)
Realistisch adressierbarer Markt bzw. Serviceable Obtainable Market (SOM)
= 74 Millionen x 0,19 x 1,34 x 200 € x 0,1 = 376.808.000 €
(376 Millionen Euro)
Dies ist eine vereinfachte Kalkulation für den gesamten Adipositas-DiGA Markt. Als DiGA-Unternehmen wird man sich meist auf spezifische ICD-Codes im Zielmarkt konzentrieren und die Rechnung kann so niedriger für die Unterauswahl der Indikationen ausfallen.
Beispiel-Rechnung für den Umsatz (Zanadio)
Wenn man sich im Adipositas-Bereich mit Zanadio ein Beispiel aus der Praxis aussieht, fallen die Umsätze zum aktuellen Zeitpunkt natürlich noch deutlich unter den beispielhaft kalkulierten, adressierbaren Markt:
Eingelöste Freischaltcodes im Zeitraum für Zanadio: 56.000
(56 Tsd – Daten des GKV-Spitzenverbandes gem. § 33a Abs. 6 SGB V; Stand 30.09.2023)
Vergütungsbetrag für Zanadio: 218,00 €
Mit diesen Werten kommt man auf diesen geschätzten Gesamtumsatz:
Umsatz = 56.000 x 218 € = 12.208.000 €
(etwa 12 Millionen Euro)
Da Zanadio zu Beginn auf Erprobung in das DiGA-Verzeichnis aufgenommen wurde, wird der reale Umsatz etwas höher liegen. Vor Verhandlung des finalen Vergütungsbetrags wurde ein Herstellerpreis von 499,80 € abgerufen. Die Kalkulation soll hier nur der Veranschaulichung dienen.
2. Kosten für die Entwicklung und Vermarktung einer DiGA
Nachdem wir nun berechnet haben, was eine DiGA maximal an Ertrag erwirtschaften kann, interessiert uns im nächsten Schritt natürlich, wie hoch die Kosten für die Entwicklung einer DiGA sind.
Die Entwicklung einer DiGA ist ein komplexer Prozess. Die Kosten können stark variieren und hängen von mehreren Faktoren ab. Um Ihnen einen besseren Überblick zu geben, listen wir in unserem kostenlosen Planungstool zur Finanzierung Ihrer DiGA alle Punkte auf, die bei der Kalkulation besonders ins Gewicht fallen.
3. Download: Excel-Planner zur Prognose von Umsatz & Kosten Ihrer DiGA
Mit unserem Excel-Planner kalkulieren Sie in wenigen Minuten den möglichen Umsatz und die Kosten Ihrer digitalen Gesundheitsanwendung.

Hinterlassen Sie hier Ihre E-Mail-Adresse und wir schicken Ihnen den Excel-Planner zu.
4. Update 2026: Anwendungsbegleitenden Erfolgsmessung (AbEM) bei DiGA
Seit Inkrafttreten des Digital-Gesetzes (DigiG) gibt es einen weiteren Faktor, dessen man sich bei der Umsatzkalkulation einer DiGA bewusst sein sollte: die anwendungsbegleitende Erfolgsmessung (AbEM). Sie ergänzt die bisherige Logik der DiGA-Vergütung, ohne diese aktuell grundlegend zu verändern.
Die AbEM wurde mit dem DigiG und der 2. Änderungsverordnung zur DiGAV (2. DiGAV-ÄndV) eingeführt und gilt für alle dauerhaft im DiGA-Verzeichnis gelisteten DiGA, die mindestens einen medizinischen Nutzen als positiven Versorgungseffekt nachgewiesen haben. Die verpflichtende Datenerhebung beginnt ab dem 01.07.2026. Die Ergebnisse sind halbjährlich an das BfArM zu melden, erstmals zum 15.04.2027.
Grundsätzlich kann die AbEM Einfluss auf die Vergütung einer DiGA haben, da gemäß § 134 SGB V ab dem 01.01.2026 ein erfolgsabhängiger Vergütungsanteil von mindestens 20 Prozent vorgesehen ist. Wie genau dieser Anteil auf Basis der AbEM-Ergebnisse berechnet wird, also ob er im Einzelfall eher erhöhend oder reduzierend wirkt, ist derzeit jedoch noch nicht abschließend geregelt.
Für die Praxis bedeutet das: Die AbEM hat potenziellen Einfluss auf die langfristige Vergütung, ist aber für die aktuelle Geschäftsmodell- und Umsatzkalkulation noch nicht konkret quantifizierbar. Für erste Wirtschaftlichkeitsrechnungen kann sie daher vernachlässigt werden, sollte jedoch bei der langfristigen Planung und Weiterentwicklung einer DiGA mitgedacht werden.
5. Fazit
Dieser Artikel hat Ihnen hoffentlich geholfen zu verstehen, wie Sie das Potenzial Ihrer DiGA-Idee abschätzen können.
Wichtig ist hierbei natürlich zu verstehen, dass wir nur auf eine Unterauswahl wichtiger Kennzahlen für die Evaluation eines DiGA-Business-Cases eingegangen sind. Es gibt eine Vielzahl an Kennzahlen, die zur Evaluation von Produkt-Ideen und Unternehmen herangezogen werden können. Zur Ihnen einen Überblick zu verschaffen haben wir uns erstmal auf die finanziellen Aspekte fokussiert, die am Anfang wohl am wichtigsten sind.
Außerdem relevant ist die Tatsache, dass die meisten Unternehmen einen Multi-DiGA-Ansatz planen und auf Basis der ersten DiGA weitere Indikationsgebiete angehen. Der Umsatz aller Indikationsgebiete sollte in diesem Fall aufsummiert werden.
Lohnt sich Entwicklung einer DiGA also schlussendlich? Wie sich über den DiGA-Excel-Planner leicht berechnen lässt, hängt dies stark von der Zielindikation Ihrer DiGA ab. Für Krankheiten mit einer hohen Prävalenz und z.B. einer digital-affinen Zielgruppe können perspektivisch beachtliche Umsätze generiert werden. Das hängt aber am Ende natürlich auch stark davon ab, ob das Marketing & der Vertrieb der gelisteten DiGA richtig geplant und ausgeführt wird (mehr dazu in unserem DiGA Marketing & Vertrieb-Leitfaden).
Die Monetarisierung Ihrer Gesundheitslösung kann auch über einen Selektivvertrag mit einer Krankenkasse erfolgen – als Alternative oder sogar Ergänzung zur DiGA. Alles, was Sie dazu wissen müssen, erfahren Sie in unserem Whitepaper.
Planen Sie eine DiGA?
Dann kontaktieren Sie uns gerne. Wir sind auf die auftragsbasierte Umsetzung von DiGA und Software-Medizinprodukten spezialisiert. Wir helfen Ihnen bei der Planung, Konzeption, der technischen Implementierung und der Einhaltung aller regulatorischen Anforderungen.
The post DiGA Umsatz & Kosten: Lohnt sich eine DiGA? appeared first on QuickBird Medical.
]]>The post Leitfaden DiGA-Marketing und Vertrieb appeared first on QuickBird Medical.
]]>Der Eintrag in das zentrale DiGA-Verzeichnis selbst führt entgegen manchem Wunschdenken nicht dazu, dass Ärztinnen und Ärzte Ihre DiGA auch verschreiben. Daher ist die Wahl der richtigen Marketing- und Vertriebsinstrumente von entscheidender Bedeutung, um das volle Marktpotenzial der jeweiligen Nische auszuschöpfen. Die Möglichkeiten sind vielfältig, das Marktumfeld komplex. Dieser Artikel soll Ihnen einen Überblick verschaffen, und hilfreiche Einordnungen auf den Weg geben.
Überblick
- 1. Patient oder Arzt – zwei mögliche Adressaten, die unterschiedlich erreicht werden
- 2. Verschiedene Kanäle stehen für verschiedene Zielgruppen zur Verfügung
- 3. Praxis-Beispiel: Faxwerbung an Arztpraxen – ein prominenter DiGA-Case
- 4. Rezeption – viele Wege führen (nicht) zum Ziel
- 5. Bedürfnisse von Patienten und Ärzten verstehen
- 6. Fazit: Verstehen und Erreichen der Zielgruppe ist der Schlüssel zum Erfolg
1. Patient oder Arzt – zwei mögliche Adressaten, die unterschiedlich erreicht werden
Die erste Grundsatzfrage einer jeden Marketingstrategie ist die nach dem Adressaten. Im Falle einer DiGA kommen zwei Gruppen infrage:
- Der Patient, der selbst nach Behandlungsmöglichkeiten im Allgemeinen oder einer auf die eigenen Bedürfnisse zugeschnittenen digitalen Anwendung im Speziellen sucht, und diese bei der Krankenkasse selbst beantragen kann, oder
- Der Arzt oder Therapeut, der die DiGA als sinnvolle Ergänzung einer Therapie erachtet, und sie dem Patienten daher verschreibt.
Der Weg vom DiGA-Hersteller zum Patienten
2. Verschiedene Kanäle stehen für verschiedene Zielgruppen zur Verfügung
Der Arzt ist laut Ernst and Young (EY) mit 90% der Fälle der häufigere Verschreibungsweg, und in einigen Fällen auch in Form einer Ersteinschätzung verpflichtend vorgeschaltet. Das „Kräfteverhältnis“ zwischen beiden Zielgruppen für DiGA-Marketing und -Vertrieb erscheint zum aktuellen Zeitpunkt eindeutig: Für die meisten DiGA-Anbieter wird sich vor allem der Vertriebs/Marketing-Fokus auf den Arzt als Verschreiber der DiGA lohnen.
Dennoch lohnt sich der Blick auf beide Zielgruppen und ihre Erreichbarkeit. Dieser holistische Blick soll Gegenstand des folgenden Abschnittes sein.
2.1 Marketing für den Patient als Zielgruppe
Eine DiGA ist in aller Regel ein Nischenprodukt. Diese klare Abgrenzbarkeit der Zielgruppe(n) kann man sich als DiGA-Anbieter insbesondere durch digitale Werbung effektiv zunutze machen.
Eine Übersicht über denkbare Marketingkanäle, die die Patienten direkt erreichen:
- Suchmaschinenwerbung- und Optimierung – Durch die Optimierung der eigenen Internetpräsenzen für die Sichtbarkeit in Suchmaschinen (SEO), besonders aber durch das Schalten bezahlter Anzeigen, die den Usern großer Suchmaschinen beim Suchen bestimmter Schlüsselbegriffe (=Keywords) ausgespielt werden, kann eine äußerst granular definierte Zielgruppe sehr zielgerichtet erreicht werden. Die erzielte Reichweite hängt in erster Linie vom eingesetzten Budget ab, und kann im Voraus relativ zuverlässig kalkuliert werden. Eher unkalkulierbar insbesondere bei neuen Produkten ist hingegen die Conversion Rate, also die (relative) Zahl der aus der Reichweite heraus tatsachlich getätigten Kaufhandlungen.
Im Falle von DiGA ist denkbar, mit dieser Methodik besonders solche Patienten zu erreichen, die sich aktiv über ihre Beschwerden, Diagnosen und denkbare Behandlungsoptionen informieren – dabei aber nicht zwangsläufig an eine DiGA denken. Als Keywords können hier beispielsweise Beschwerden und Diagnosen gesetzt werden. - Social-Media-Marketing – Die sozialen Medien spielen als Informations- und Werbeplattform inzwischen eine entscheidende Rolle. Entsprechend sinnvoll könnte es sein, mit der eigenen DiGA auch hier Präsenz zu zeigen. Im Allgemeinen sind die Erfolgsfaktoren ein authentischer Auftritt, regelmäßiger Content und die Wahl der richtigen Kanäle. Die gewählten Plattformen müssen ebenso zum Produkt und der Zielgruppe passen wie die Art des Auftritts, die Bildsprache, das Wording und der Content. Eine Zielgruppe 50+ wird anders angesprochen als die Generation Z. Der Contentmix für die Bewerbung einer DiGA kann denkbarerweise aus abwechselnden Produktinformationen und Testimonials bzw. Erfahrungen von Nutzern bestehen.
Auch zwei Spezialitäten von Social Media sind als Teil des Marketingmix‘ theoretisch denkbar: Die Zusammenarbeit mit Influencern (wenn diese als Teil der Zielgruppe glaubwürdig die DiGA bewerben können) und das Schalten von Werbekampagnen. (An dieser Stelle sei darauf hingewiesen, dass bei der Erstellung von Werbematerialien, Texten etc., die sich hier an Patienten direkt richten, besondere Sorgfalt geboten ist. Da eine DiGA immer auch ein Medizinprodukt ist, gilt hier das Heilmittelwerbegesetz (HWG). Beispielsweise darf keinesfalls suggeriert werden, dass der Einsatz der DiGA mit Sicherheit erfolgreich sein wird. Allzu reißerische Formulierungen sind also auch auf Social Media tabu, sonst drohen empfindliche Strafen. - Display-Advertising – Hier werden auf i.d.R. gut frequentierten Webseiten (z.B. Nachrichtenportalen) frei gestaltbare Werbeanzeigen zwischen den eigentlichen Inhalten angezeigt. Auch hier lässt sich bei den meisten Anbietern (hier ist die Auswahl größer als bei den Suchmaschinen oder sozialen Medien) gut filtern, welche Personen mit welchen Interessen und welchen demografischen Merkmalen die Anzeige sehen sollen. Die Kosten hängen auch hier von der erzielten Reichweite ab – der Streuverlust ist hier jedoch relativ hoch. Im speziellen Fall einer DiGA kommt hinzu, dass diese hier mit zahlreichen anderen beworbenen Produkten um ein begrenztes Maß an Aufmerksamkeit konkurriert.
- Klassische Massenmedien – Natürlich besteht auch für digitale Produkte die Möglichkeit, diese in klassischen, analogen Kanälen (TV, Radio, Print…) zu bewerben. Verschiedene Punkte sprechen jedoch eher gegen diesen Weg – im Gegensatz zu allen digitalen Kanälen besteht außer der Auswahl des Mediums selbst keinerlei Möglichkeit, das Publikum zielgerichtet zu filtern, ebenso fehlen Instrumente zur Erfolgsmessung und Ergebnisanalyse. Die Investitionen in die Produktion der Medien selbst (Radiospots, TV-Clips…) sind sehr hoch, gleiches gilt für deren Ausspielung. Dem gegenüber steht ein hoher Anteil „toter“ Reichweite.
- (Werbe-)Partnerschaft mit Krankenkassen – Theoretisch ist die Kooperation mit Krankenkassen auch ein denkbarer Weg, Patienten direkt zu erreichen. Schließlich stellen diese die Schnittstelle zwischen Patienten und dem Gesundheitssystem dar, und verfügen über ein weit verzweigtes Kommunikationsnetz zu den Versicherten. Fraglich ist allerdings, wie diese von einer Kooperation überzeugt werden sollen: Werbewirkung für die Krankenversicherung selbst können sich zumindest die gesetzlichen Krankenkassen nicht (mehr) erhoffen, da sie durch den Gesetzgeber sowieso zur Erstattung der DiGA verpflichtet sind.
2.2 Vertrieb & Marketing für den Arzt als Zielgruppe
Die mit 90% der Verschreibungen deutlich wichtigere Zielgruppe stellt die Ärzteschaft dar. Diese kann besonders auf Wegen erreicht werden, die sich im Bereich des DiGA-Marketings/-Vertriebs von solchen des klassischen Pharmavertriebes wenig unterscheiden.
Hinweis: Aus Gründen der Lesbarkeit wurde in diesem Text der Begriff „Arzt“ als Synonym für alle Mediziner und Therapeuten verwendet, die DiGA verschreiben dürfen.
- Direktvertrieb: Klassischer Außendienst – Für Ärzte ist es Gewohnheit, regelmäßig von Außendienstlern der Pharmabranche aufgesucht zu werden. Durch den persönlichen Kontakt zu einem festen und physisch greifbaren Ansprechpartner wird Vertrauen und damit eine Bindung geschaffen. Auch ein DiGA-Hersteller kann prinzipiell diesen Weg gehen, um eine Anwendung in der Ärzteschaft zu etablieren. Der Aufbau eines kompletten Außendienstnetzes ist fraglos mit nicht unerheblichen Kosten verbunden. Dass er sich jedoch durchaus amortiseren kann, hat Marcus Bergler, Experte für DiGA-Vertrieb, in seinem Artikel plausibel errechnet. Dieser Vertriebskanal kommt der Ärzteschaft mutmaßlich am weitesten entgegen, da sie ihn bereits aus der Pharmabranche kennt.
- Direktvertrieb: Zusammen mit Partnern – In diesem Kontext ist zudem ein interessanter Trend zu beobachten: DiGA-Start-Ups schließen sich in unterschiedlichen Formen mit Pharmaunternehmen zusammen, Beispiele sind hier die Kooperationen von Kalmeda mit Pohl-Boskamp oder HelloBetter und Ratiopharm. Auch Pfizer und Roche zeigen großes Interesse am DiGA-Markt. Die bestehende, etablierte und weit verästelte Vertriebsstruktur der Pharmaunternehmen kann für den Vertrieb der DiGA mitgenutzt werden, im Gegenzug können die Pharmaunternehmen vom technologischen Know-How der Start-Ups profitieren. Eine Alternative zu diesem Modell – für solche DiGA-Hersteller, die von der Pharmaindustrie unabhängig agieren wollen – stellt eine gegenseitige Kooperation dar. DiGA-Hersteller bauen ein gemeinsames Vertriebsnetz auf, teilen sich somit Kosten und Risiken, und stellen bestmögliche Auslastung sicher.
- Klassische Breitenwerbung – Während der oben beschriebene „klassische“ Direktvertrieb wohl das zentrale Instrument zum unmittelbaren Vermarkten des Produktes ist, kann dieses durch flankierende Maßnahmen mit dem Zweck der Bekanntheitssteigerung und des allgemeinen Imagebuildings unterstützt werden. Klassische Mittel sind hier z.B. Werbeanzeigen oder optimalerweise das Erreichen „echter“ Berichterstattung in Fachmedien ebenso wie das Versenden speziell für die Ärzteschaft konzipierter Broschüren an Praxen mit relevantem Schwerpunkt. Ähnlich wie in anderen Branchen auch finden für Ärzte zudem regelmäßig Events statt. Je nach Format kann es sinnvoll sein, durch z.B. einen Stand und/oder einen Fachvortrag Präsenz zu zeigen.
- Sponsorings von Fortbildungen – Eine subtilere Maßnahme zur Bekanntheitssteigerung kann die Übernahme eines Sponsorings sein: Jeder Facharzt ist verpflichtet, innerhalb von fünf Jahren 250 sogenannter CME-Punkte zu erwerben. Die entsprechenden Fortbildungen werden häufig kostenfrei angeboten, und finanzieren sich u.A. über Sponsorings, z.B. von Arzneimittelherstellern. Auch wenn ein Sponsoring keinerlei Einfluss auf die Inhalte der Fortbildung haben darf, erhält der Sponsor doch eine gewisse Präsenz in der Veranstaltung, sei es durch Erwähnung und Positionierung des Namens oder Logos oder durch die Auslage von Werbeartikeln. Es existieren bereits CME-Fortbildungen speziell zum Thema DiGA. Noch geschickter kann sein, solche Fortbildungen zu sponsern, die sich mit dem durch die DiGA adressierten Krankheitsbild befassen. Mit einer solchen Maßnahme besteht Zugang zu einem Publikum, das durch die DiGA fachlich tangiert wird, und zugleich ggf. durch das Sponsoring erstmals von einer solchen hört.
Hinweis: Die Liste der genannten Distributionskanäle ist sicher nicht vollständig und sollen Ihnen vor allem einen initialen Überblick über einige möglich Maßnahmen geben. Sie sollten grundsätzlich auch andere Wege in Betracht ziehen, die hier nicht explizit genannt wurden.
3. Praxis-Beispiel: Faxwerbung an Arztpraxen – ein prominenter DiGA-Case
Ein viel beachteter Fall aus dem DiGA-Markt betrifft einen Hersteller einer Adipositas-DiGA, der eine gezielte Marketingtaktik einsetzte: Patient:innen, die Interesse an der DiGA zeigten, wurden vom Hersteller aktiv dazu bewegt, ihm eine Vollmacht zur Kontaktaufnahme mit der behandelnden Hausarztpraxis zu erteilen. Auf dieser Grundlage versandte der Hersteller ein Fax an den Arzt, um die Ausstellung einer Verordnung oder eines Kurzattestes anzustoßen.
Das Fax enthielt ein Anschreiben, ein Formular für ein Kurzattest (in dem die DiGA bereits voreingetragen war) sowie ein Formular zur kostenlosen Anforderung weiterer Informationen zur DiGA. Zudem wies der Hersteller auf abrechenbare ärztliche Leistungen im Zusammenhang mit der Nutzung der DiGA hin. Der Ansatz erwies sich als sehr wirksam und führte kurzfristig zu einer hohen Zahl an Verordnungen.
Rechtlich ist dieser Vertriebsweg jedoch nicht zulässig. Mit Urteil vom 18.11.2025 (Az. 6 U 130/24) stellte das OLG Brandenburg klar, dass es sich hierbei um unzulässige Werbung per Telefax nach § 7 UWG handelt, da der Arzt in eine solche Werbung nicht eingewilligt hatte. Maßgeblich war, dass das Fax über eine neutrale Weiterleitung des Patientenanliegens hinausging und gezielt absatzfördernde Elemente enthielt.
Der Fall zeigt exemplarisch: Auch sehr effektive, patientenseitig initiierte Marketingtaktiken stoßen im DiGA-Vertrieb an klare rechtliche Grenzen.
4. Rezeption – viele Wege führen (nicht) zum Ziel
Dass das Thema DiGA für den angesprochenen Arzt Neuland ist, ist nicht unwahrscheinlich. Laut einer Umfrage des Fraunhofer-Instituts aus dem Jahr 2021 „schätzen 75% der teilnehmenden Ärzte ihren Informationsstand zu DiGA als schlecht oder sehr schlecht ein“. Entsprechend hoch ist der Informationsbedarf und das Interesse: In derselben Umfrage erklären 37% der Ärzte eine „hohe oder sehr hohe Bereitschaft“, DiGA zu verordnen. Bisher getan haben dies laut eigenen Angaben jedoch erst 2%. Dennoch machen, wie erwähnt, Verschreibungen durch Ärzte 90% des Gesamtmarktes aus.
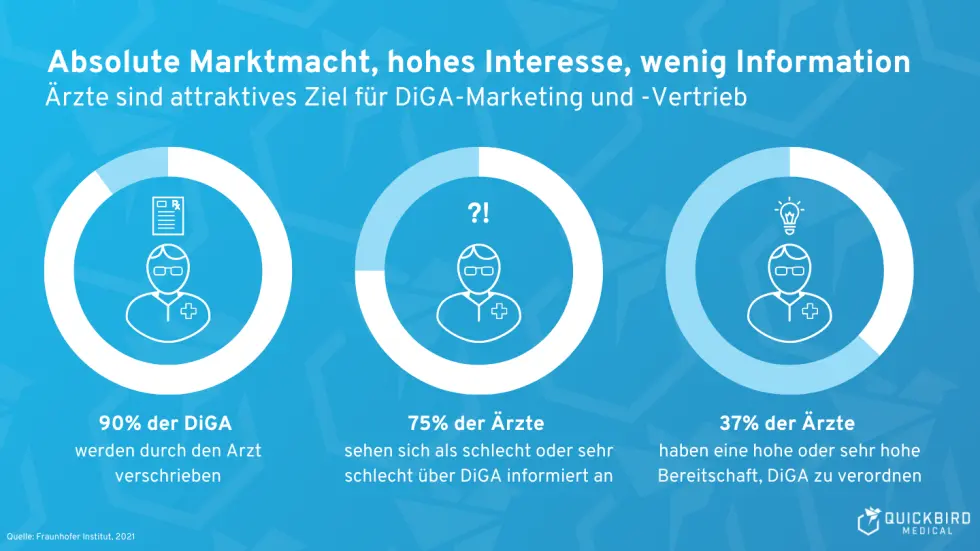
Ärzte sind ein attraktives Ziel für DiGA-Marketing
Geringer Informationsgrad, hohes Interesse, absolute Marktmacht: Die Ärzteschaft als Zielgruppe ist somit die „Low Hanging Fruit“, die es als ersten Schritt nach einem Markteintritt zu ernten gilt. Naheliegendster und erfolgversprechendster Schritt ist, hierfür den direkten Kontakt zu suchen. Kann oder will man das als einzelner DiGA-Hersteller nicht allein bewältigen, bietet sich an, mit Pharmaunternehmen oder anderen DiGA-Herstellern zu kooperieren. Flankiert werden kann das durch diverse Maßnahmen zur Steigerung des Bekanntheitsgrades: Werbeaktionen, Präsenzen auf Kongressen und Veranstaltungen oder Sponsoring entsprechender Fortbildungen.
Mit dem steigenden Bewusstsein für Gesundheit in der Gesellschaft einserseits und dem wachsenden Bekanntheitsgrad digitaler Technologien andererseits ist denkbar, dass sich das schnell ändert. Gegebenenfalls induziert durch Marketing- und Vertriebsaktivität seitens der DiGA-Hersteller selbst. Zur Vermarktung einer DiGA an den Patienten direkt steht nahezu die gesamte Klaviatur der Produktwerbung zur Verfügung, mit Blick auf die sehr klare Abgrenzbarkeit und die vorausgesetzte Digitalaffinität der Zielgruppe(n) erscheint besonders solche in digitaler Form sinnstiftend. Das Risiko ist hier zugleich geringer: Fixkosten entstehen nicht.
5. Bedürfnisse von Patienten und Ärzten verstehen
Ein Schlüssel für die erfolgreiche Definition der eigenen Marketingstrategie ist neben der Wahl der Kanäle auch die der Inhalte. Für beides von Bedeutung ist das Verstehen der Denkweise und Bedürfnisse der Zielgruppen. “Der Erfolg des digitalen Wandels im Gesundheitswesen steht und fällt damit, wie weit Patienten, Versicherte und Leistungserbringer digitale Lösungen akzeptieren”, stellt die Bertelsmann-Stiftung fest.
Der Markt muss nach der entsprechenden DiGA „verlangen“. Die Zielgruppe „Patienten“ muss eine sein, die eine gesundheitliche Herausforderung mit hohem individuellen Leidensdruck und zugleich begrenzten Therapiemöglichkeiten mit sich bringt. An diesen Punkten kann und muss das DiGA-Marketing den Patienten „abholen“, und ihm verdeutlichen, dass mit der DiGA eine unkonventionelle Methode zur Verfügung steht, die zudem bestenfalls äußerst nutzerfreundlich in den Alltag zu integrieren ist. Wie Sie Ihre Zielgruppe zielführend definieren und einen positiven Versorgungseffekt nachweisen, erfahren sie in unserem Leitfaden.
Für die Zielgruppe „Ärzteschaft“ ist von Bedeutung, dass die DiGA nachweislich „funktioniert“. Ärzte hören z.B. auch auf das Feedback, das Patienten diesen Nach Nutzung übermitteln. Wenn der Patient nicht von der DiGA überzeugt ist oder diese nicht nutzt, sinkt die Wahrscheinlichkeit für Folgeverordungen drastisch.
6. Fazit: Verstehen und Erreichen der Zielgruppe ist der Schlüssel zum Erfolg
Die Vielfalt an denkbaren Marketing- und Vertriebswegen ist enorm. Das kann besonders zum Start erschweren, sich für einen Ansatzpunkt, eine Strategie (und bestenfalls die „richtige“) zu entscheiden.
Klar ist jedoch auch: Eine universelle Handlungsempfehlung kann es nicht geben. Jede DiGA ist ein einzigartiges Produkt in einem jeweils einzigartigen und komplexen Marktumfeld. Für einen erfolgreichen Marktstart ist essenziell, dieses Umfeld genau zu analysieren, und darauf aufbauend die Marketing- und Vertriebsmethodik zu wählen, die am besten zu Produkt und Zielgruppe passt.
Sie stehen momentan noch am Anfang? Welche Faktoren Sie bei der Planung und Umsetzung Ihrer DiGA beachten sollten, erfahren Sie in unserem Whitepaper „13 Empfehlungen für die Zulassung von DiGA in 2026“.
Mit diesem Artikel war unser Ziel, Ihnen einen breit gestreuten Überblick über denkbare Instrumente zu verschaffen, und diese, der Natur eines Blogartikels entsprechend, verallgemeinert einzuordnen.
Über uns: QuickBird Medical ist auf die Umsetzung von DiGA auf Auftragsbasis spezialisiert. Falls Sie eine DiGA planen, sprechen Sie uns gerne an: [email protected].
The post Leitfaden DiGA-Marketing und Vertrieb appeared first on QuickBird Medical.
]]>The post Software als Medizinprodukt: 13 Zertifizierungs-Tipps für Hersteller appeared first on QuickBird Medical.
]]>1. Priorisieren Sie einen zeitnahen Marktstart
Gewinnbringende Geschäftsmodelle im Gesundheitswesen zu etablieren, erfordert Zeit. Wenn Sie z.B. Krankenkassen oder potenzielle B2B-Kunden von Ihrer Software-Lösung überzeugen möchten, werden Sie ggf. viel Zeit in Besprechungen und Verhandlungen verbringen. Dies kann sich Monate oder sogar Jahre in die Länge ziehen. Je später Sie mit diesen Erstkontakten beginnen, desto später können Sie mit Ihrer Lösung Umsatz generieren. Um in diese Besprechungen einzusteigen, benötigen Sie jedoch meistens schon einen funktionierenden Prototyp (bzw. MVP) oder sogar ein zertifiziertes Software-Medizinprodukt inklusive wissenschaftlicher Evidenz über den medizinischen Mehrwert und/oder Kosteneinsparungspotenziale. Daher sollten Sie sich in der Planung Ihrer Software-Features nicht verlaufen. Fokussieren Sie sich auf die Kernfunktionen und setzen Sie erstmal nur diese Kernfunktionen um. Alle Erweiterungen, die nicht unbedingt nötig sind („Nice-To-Have“), sollten Sie eventuell erst später umsetzen. Um dies nochmal zu betonen: Je mehr Zeit Sie für die Planung und Umsetzung der ersten Produkt-Version benötigen, desto später können Sie Umsatz am Markt generieren. Sales-Zyklen am Gesundheitsmarkt sind lang. Fangen Sie nicht zu spät damit an. Planung ist natürlich gerade bei regulierten Medizinprodukten, die einer Zulassung und Zertifizierung bedürfen, enorm wichtig. Optionale Zusatz-Features können aber auch bei Medizinprodukt-Software teilweise erstmal weglassen und auf spätere Produkt-Versionen verschoben werden.
Unsere Empfehlung: Starten Sie mit etwas Einfachem, das dennoch einen hohen Mehrwert für Ihre Zielgruppe mit sich bringt. Versuchen Sie, schnell auf den Markt zu kommen. Um Erweiterungen, weitere Apps, oder stark innovative Features können Sie sich im nächsten Schritt kümmern.
2. Nutzen Sie vorhandene wissenschaftliche Evidenz
Um Ihr Software-Medizinprodukt auf den Markt zu bringen, müssen Sie für die Zertifizierung unter anderem wissenschaftlich nachweisen, dass Ihr Produkt den angestrebten medizinischen Zweck erfüllt. Für Produkte der Risikoklasse IIa oder höher müssen Sie hierfür womöglich eine aufwendige klinische Prüfung (Studie für Ihr Medizinprodukt) durchführen. Für Produkte der Risikoklasse I (gerade Produkte, die auf existierende medizinische Leitfäden aufsetzen) reicht der Aufsichtsbehörde gegebenenfalls eine systematische Literatur-Recherche. Wenn Ihr Produkt also existierende medizinische Leitfäden oder Methoden digitalisiert, können Sie potenziell die existierende wissenschaftliche Evidenz nutzen, um Ihr Produkt zu validieren. Sie benötigen somit eventuell keine kostspielige klinische Studie bzw. klinische Prüfung. Ob dies für Ihr Produkt wirklich ausreichend ist, muss natürlich im Einzelfall abgeklärt werden. Wenn Ihr Produkt allerdings hochgradig innovativ ist und Sie komplett neue Methoden entwickeln, liegt hierfür natürlich keine Evidenz vor. Sie müssen diese Evidenz selbst generieren und z.B. eine klinische Studie durchführen. Wenn Sie triftige Gründe haben anzunehmen, dass Ihr Produkt einen medizinischen Nutzen nachweisen kann und am Markt viel Umsatz generieren wird, kann sich eine solche klinische Studie lohnen. Dies hängt vom Produkt und Anwendungsfall ab.
Unsere Empfehlung: Für den initialen Marktstart sollten Sie versuchen, erstmal existierende wissenschaftliche Evidenz zu nutzen, wenn dies möglich ist. Dies ermöglicht Ihnen potenziell einen deutlich schnelleren Marktstart ohne klinische Prüfung/Studie. Später werden Sie ggf. trotzdem weitere Studien zu Ihrem Produkt fahren (dies hilft auch ungemein bei der Vermarktung Ihres Produkts). Zu diesem Zeitpunkt ist Ihr Produkt aber im Optimalfall bereits am Markt und generiert Umsatz – eine deutlich angenehmere Ausgangslage.
3. Visieren Sie Risikoklasse I an
Eine Zeit lang kursierte die Nachricht, dass es unter MDR kaum noch Software-Medizinprodukte der Risikoklasse I geben würde, was zum Glück nicht stimmt. Fast alle unter MDR zugelassenen Anwendungen im DiGA-Verzeichnis sind der Risikoklasse I zugeordnet. Auf unserem DiGA-Dashboard für Hersteller erhalten Sie einen genauen Überblick über die Verteilung der Risikoklassen bereits gelisteter DiGA. Ob Ihr Medizinprodukt in die Risikoklasse I fällt, hängt von verschiedenen Aspekten ab. Werfen Sie einen Blick in unseren Leitfaden zur Klassifizierung von Software-Medizinprodukten nach MDR, um dies herauszufinden. Normalerweise ist die Zertifizierung von Software der Risikoklasse IIa mit einem erheblich höheren Aufwand und zusätzlichen Kosten verbunden, da eine benannte Stelle eingeschaltet werden muss. Bei der Risikoklasse I haben Sie weitaus höhere Chancen, Ihr Produkt auch ohne eine klinische Prüfung zuzulassen. Es könnte sich daher lohnen, Ihre Anwendung so aufzusetzen, dass sie in die Risikoklasse I fällt. Wir verstehen jedoch, dass solche Änderungen am Konzept nicht für jedes Produkt möglich sind. Insbesondere bei Produkten, die von Patienten verwendet werden, kann es jedoch häufig funktionieren. Es lohnt sich also möglicherweise, darüber nachzudenken. Gerne beraten wir Sie zu diesem Thema und evaluieren, ob die Risikoklasse I für Ihr Produkt geeignet ist.
Unsere Empfehlung: Gestalten Sie Ihre Anwendung so, dass eine Klassifizierung nach Risikoklasse I möglich ist, um erheblichen Mehraufwand ab Risikoklasse IIa zu vermeiden.
4. Wählen Sie die richtige Zielgruppe
Die gewählte Zielgruppe und der Verwendungszweck Ihrer Medizinprodukt-Software bestimmt maßgeblich über den Erfolg Ihrer Unternehmung. Relevante Fragen, die Sie evaluieren sollten, könnten sein:
| Frage | Beispiel |
| Ist diese Zielgruppe digital affin? Gibt es eine ähnliche Zielgruppe, die digital-affiner wäre? | Wenn das Produkt von pflegebedürftigen Personen verwendet werden soll, ist das Durchschnittsalter der Zielgruppe vermutlich verhältnismäßig hoch. Die Zielgruppe ist daher weniger digital-affin als z.B. eine 20-jährige Patientin. Welche besonderen Bedürfnisse gilt es zu beachten? Könnten Sie stattdessen die Angehörigen der pflegebedürftigen Personen adressieren? Denn das sind oft junge Personen, die z.B. ihren pflegebedürftigen Eltern helfen und finanzielle Entscheidungen für diese treffen. |
| Adressieren Sie einen vorhandenen großen Schmerzpunkt Ihrer Zielgruppe oder ist Ihre App nur ein nice-to-have für diese Zielgruppe? | Viele Patienten mit mentalen Problemen haben mit langen Wartezeiten für Psychotherapeuten zu kämpfen. Diese Wartezeit stellte bisher eine Versorgungslücke dar, die nun viele Startups im Bereich Mental Health versuchen zu schließen. Der Leidensdruck der Zielgruppe ist groß und die Zielgruppe größtenteils digital affin, weshalb die Zielgruppe potenziell hohes Interesse an einer digitalen Anwendung hat. |
| Ist der Arzt oder der Patient Ihre Hauptzielgruppe? | Einem Produkt, das nur funktioniert, wenn sowohl Patienten und Ärzte an Bord sind, stehen große Hürden beim Marktzugang bevor. Sie müssen bei beiden Zielgruppen eine kritische Masse an Menschen erreichen. Fokussieren Sie sich daher lieber (falls möglich) erstmal nur auf eine der beiden Parteien und bauen Ihr Produkt so auf, dass es auch ohne Mitwirken der anderen Partei funktionieren könnte. Später können Sie Stück für Stück auch die andere Partei in Ihre Lösung involvieren. |
Dies sind nur einige der relevanten Punkte bei der Wahl der Zielgruppe, aber es soll Ihnen ein Verständnis geben, wie wichtig die präzise Definition der richtigen Zielgruppe für Ihr Produkt ist.
Unsere Empfehlung: Führen Sie qualitative Interviews durch, recherchieren Sie zusätzliche Daten und testen Sie Ihr Produkt mithilfe eines Click-Dummies an Ihrer Zielgruppe. Somit können Sie frühzeitig erkennen, ob Sie die richtige Zielgruppe gewählt haben bzw. ob das Produkt zur Zielgruppe passt und vermeiden kostspielige Strategiewechsel im späteren Produktverlauf.
5. Nehmen Sie Datensicherheit und Datenschutz ernst
Die Datensicherheit und der Datenschutz laufen Gefahr, während der Produktentwicklung zu niedrig priorisiert zu werden. Dies liegt auch daran, dass der Nutzen von Datensicherheit leider erst dann klar wird, wenn es schon zu spät ist und z.B. ein Angreifer die Daten Ihrer Patienten gestohlen hat. Ein solches Datenleck kann neben möglichen Strafzahlungen einen massiven Reputationsschaden an Ihrem Unternehmen und Produkt anrichten. Dies sollten Sie natürlich auf das Nötigste vermeiden.
Leider bringt die Umsetzung des Datenschutzes nach DSGVO und der Datensicherheitsmaßnahmen z.B. nach ISO/IEC 27001 und BSI-Standards einen entsprechenden Aufwand mit sich. Sie brauchen Experten, die sich in diesem Gebiet auskennen und einen strukturierten Ansatz, um nicht nur technische, sondern auch organisatorische Datensicherheits-Risiken zu mitigieren. Hierfür gibt es existierende Leitfäden und Standards, die Sie nutzen sollten (z.B. Technische Richtlinien vom BSI, Leitfäden der OWASP Foundation und Normen wie die ISO/IEC 27001 und die IEC 81001-5-1).
Wichtig ist hierbei vor allem: Auch um die Kosten Ihrer Software oder App durch Krankenkassen erstatten zu können, ob über den DiGA-Pfad oder Selektivverträge, werden bestimmte Zusicherungen der Datensicherheit notwendig. DiGA müssen sogar eine Zertifizierung nach BSI TR-03161 vorweisen.
Unsere Empfehlung: Nehmen Sie das Thema Datensicherheit und Datenschutz sehr ernst. Bauen Sie intern Cyber-Security- und Datenschutz-Kompetenz auf oder arbeiten nur mit Dienstleistern, die nachweislich Datensicherheits-Prozesse (z.B. ein Information Security Management System nach ISO/IEC 27001 und IEC 81001-5-1) etabliert haben und DSGVO-Konformität nachweisen können.
6. Machen Sie sich ein holistisches Bild über verschiedene Finanzierungsoptionen
Je nachdem, ob Sie eine Patienten-App oder eine Software für Ärzte entwickeln, haben Sie viele verschiedene Finanzierungs-Optionen, die auch mit unterschiedlichen Zertifizierungs-Wegen einhergehen. Eine Software für Healthcare Professionals könnte z.B. einzeln an Arztpraxen oder Kliniken verkauft werden, Fördermittel des Bundes nutzen, den Weg über Hilfsmittel-Verzeichnis des GKV gehen oder durch Krankenkassen finanziert werden. Eine Patienten-App könnte z.B.
- … durch Selektivverträge mit Krankenkassen erstattet werden.
- … als DiGA im Rahmen des Digitale-Versorungs-Gesetzes finanziert werden
- … von der Zentrale Prüfstelle für Prävention (ZPP) zertifiziert und so über Krankenkassen erstattet werden.
- … im Selbst-Zahler-Modell durch Patienten bezahlt werden.
Die Entscheidung für die richtige Option sollten Sie mit Bedacht treffen. Hier ist z.B. eine Gegenüberstellung zweier Optionen für die Erstattung einer Patienten-App durch Krankenkassen:
| Finanzierung über Selektivverträge mit Krankenkassen | Finanzierung über Digitale-Versorgung-Gesetz (DiGA-Fast-Track) |
| Jede Krankenkasse muss separat überzeugt werden | Sofortiger Zugriff auf Erstattung durch alle gesetzlichen Krankenkassen |
| Keine transparenten, standardisierten Prüf-Kriterien | Standardisierter, transparenter Prüfprozess |
| Fokus ist meist auf möglichen Kostenreduktionen für die Krankenversicherung | Fokus kann auf Verbesserung der menschlichen Gesundheit liegen und ist dabei erstmal unabhängig vom Kosteneinsparungspotenzial für die Krankenkassen |
| Wissenschaftliche Evidenz muss meist vor dem Erstattungszeitpunkt vorliegen | Potenzial wissenschaftlicher Evidenz muss durch Evaluationskonzept (und Pilotstudie) initial geprüft werden, aber es muss im ersten Jahr keine vollumfängliche klinische Studie vorliegen |
| Kein klarer Zeitrahmen zur Finanzierung | Klarer Zeitrahmen durch gesetzlich festgeschriebene Prüfungsfristen (Rückmeldung innerhalb von 3 Monaten etc.) |
| Anforderungen an Studien sind ggf. niedriger | Sehr hohe Anforderungen an das Studien-Design |
| Anforderungen an Ihre Anwendung sind ggf. niedriger | Hohe Anforderungen an Datensicherheit, Datenschutz, Barrierefreiheit, Interoperabilität etc. |
| Anwendungsfälle denkbar, die rechtlich nicht als DiGA zugelassen werden können (z.B. Primärprävention) | DiGA sind beschränkt auf Patienten-zentrierte Anwendung, die meist der Therapie dienen (mehr dazu lesen Sie in unserem Blog-Artikel) |
Unsere Empfehlung: Evaluieren Sie vorab, welcher Finanzierungsweg am besten zu Ihrem Produkt und Ihrem Unternehmen passt. Sprechen Sie mit Experten und anderen Unternehmen, die solche Wege schon mit Ihren Produkten gegangen sind, um Erfahrungswerte zu hören. Klären Sie dies frühzeitig, weil durch das gewählte Finanzierungsmodell ggf. Zusatzanforderungen an Ihr Produkt oder die nötige wissenschaftliche Evidenz gestellt werden.
Sieben weitere Tipps zur Zertifizierung erhalten Sie in unserem Whitepaper…
Alle weiteren Zertifizierungs-Tipps finden Sie in unserem Medizinprodukt-Whitepaper. Einfach über das Formular downloaden.
Fazit: Erhöhen Sie die Erfolgswahrscheinlichkeit Ihrer Medizinprodukt-Software
Die Entwicklung einer Medizinprodukt-Software ist durch die regulatorischen Hürden kein Unterfangen, das unterschätzt werden sollte. Mit guter Planung und der richtigen Beratung können Sie die Chancen für die Zulassung und Zertifizierung und damit den erfolgreichen Markteintritt stark erhöhen. Wir hoffen, dass Ihnen die obigen Empfehlungen helfen, unnötigen Aufwand und potenzielle Risiken zu vermeiden. Weitere Informationen zu Themen wie dem Medizinprodukt-Zulassungsprozess, der Durchführung einer klinischen Bewertung für Ihre Software und sonstigen Themen im Qualitätsmanagement finden Sie auf der Übersichtsseite unseres DiGA- und Medical Software-Blogs.
The post Software als Medizinprodukt: 13 Zertifizierungs-Tipps für Hersteller appeared first on QuickBird Medical.
]]>The post 13 Tipps für die Zulassung von DiGA in 2026 appeared first on QuickBird Medical.
]]>Und tatsächlich – viele zertifizierte DiGA, die es auf den Markt schaffen, zeigen überaus positive Verordnungs- und Umsatzzahlen. Andere hingegen bekommen keine Zulassung oder scheitern bereits vor der Antragstellung. Doch warum ist es so? Welche Faktoren beeinflussen das Scheitern oder den Erfolg Ihrer DiGA? In diesem Artikel fassen wir die wichtigsten Faktoren und Empfehlungen zusammen, die Sie im Jahr 2026 bei der Planung und Umsetzung bis hin zu der Zulassung Ihrer DiGA beachten sollten.
Inhaltsverzeichnis
- 1. Der positive Versorgungseffekt
- 2. Planen Sie BSI TR-03161 bereits beim Produktkonzept mit ein
- 3. Planen Sie Ihre klinische Studie ausreichend
- 4. Arbeiten Sie mit den richtigen Medizinprodukt-Beratern zusammen
- 5. Nutzen Sie das Beratungsangebot des BfArM
- 6. Visieren Sie Risikoklasse I an
- 7. Verwenden Sie existierenden Code zur Umsetzung der ePA- und GesundheitsID-Anbindung
1. Der positive Versorgungseffekt im Fokus
Bei der Konzeption und dem Design Ihrer DiGA sollte der positive Versorgungseffekt im Mittelpunkt stehen. Wenn Ihre Studie am Ende keinen signifikanten Effekt auf die Patientenpopulation nachweisen kann, wird Ihre DiGA auch nicht dauerhaft im DiGA-Verzeichnis aufgenommen. Das sollte daher der Mittelpunkt jeglicher Produkt-Design-Entscheidungen sein.
Studien-Endpunkte frühzeitig definieren
Definieren Sie frühzeitig, welchen positiven Versorgungseffekt Sie anstreben möchten. Seien Sie hierbei so konkret wie möglich. Definieren Sie die primären und sekundären Endpunkte Ihrer Studie ggf. bereits während der Produktplanung.
Design mit Fokus auf positivem Versorgungseffekt
Jedes Produkt-Feature, das nicht unmittelbar zu einer Steigerung des angestrebten Versorgungseffekts beiträgt, sollte hinterfragt werden. Es ist ohnehin schon eine große Herausforderung, im DiGA-Verzeichnis zu landen. Jede zusätzliche Produktkomplexität erhöht Ihre Kosten und Time-to-Market. Sie sollten zusätzliche Funktionen nur einbauen, wenn diese auch sehr wahrscheinlich einen positiven Einfluss auf den Versorgungseffekt haben.
Nutzung von existierender Evidenz
Nutzen Sie beim Design existierende Evidenz in Ihrem angestrebten Indikationsgebiet. Anerkannte Leitfäden bieten eine solide empirische Grundlage und etablierte Therapiemethoden sind gut erforscht. Daher stehen auch die Chancen gut, dass Sie mit Ihrer digitalen Lösung ebenfalls einen positiven Versorgungseffekt nachweisen können.
Unsere Empfehlung:
Stellen Sie sich bei jeder Produkt-Funktion die Frage: „Hat diese Funktion einen Einfluss auf den positiven Versorgungseffekt meiner DiGA?“. Features, die „nice-to-have“ sind, sollten Sie erstmal weglassen und sich auf die medizinisch relevanten Produkt-Funktionen fokussieren.
2. Planen Sie BSI TR-03161 bereits beim Produktkonzept mit ein
Seit 1. Januar 2025 müssen DiGA zur Aufnahme in das DiGA-Verzeichnis vorher von einer akkreditierten Prüfstelle nach BSI TR-03161 zertifiziert werden.
Die Sicherheitsanforderungen der BSI-TR-03161 sind umfangreich und haben teilweise einen tiefgreifenden Einfluss auf das technische Konzept und sogar die Nutzeroberfläche Ihrer DiGA.
Eine fertig entwickelte DiGA nachträglich BSI-konform zu machen ist allerdings deutlich mehr Aufwand als die Anforderungen von Anfang an bei der technischen Planung zu integrieren.
Die Sicherheitsanforderungen der BSI TR-03161 beeinflussen z.B. indirekt:
- Die Auswahl der Software-Frameworks & -Bibliotheken
- Die technische Architektur
- Die Auswahl des Server-Anbieters (Stichwort „C5-Zertifikat“)
- Verschlüsselungskonzept
- Technische Kommunikation der Komponenten
- Authentifizierungsmethoden
- Etc.
Unsere Empfehlung:
Wenn Sie Aufwand und Kosten sparen möchten, beschäftigen Sie sich mit der BSI TR-03161 bevor Sie die Entwicklung Ihrer Software beginnen. Identifizieren Sie die Anforderungen, die maßgeblichen Einfluss auf Ihr technisches Konzept haben und integrieren Sie diese entsprechend frühzeitig. Gerne unterstützen wir Sie hierbei mit Beratungs- oder Entwicklungsleistungen (hier finden Sie mehr Informationen).
3. Planen Sie Ihre klinische Studie ausreichend
Im Startup-Bereich gibt es viele einflussreiche Paradigmen, die es Unternehmern erlauben, die Erfolgswahrscheinlichkeit der eigenen Firma zu erhöhen:
- „Move fast and break things“ (Facebook)
- „Responding to change over following a plan“ (Agile Software Development Manifesto)
- „Fail Faster“
Diese Mantren und Strategien sind in gewissen Kontexten enorm hilfreich und wichtig. Wenn es um die Planung eines Medizinprodukts oder einer DiGA geht, ist Voraus-Planung jedoch deutlich wichtiger als im klassischen Software-Startup-Bereich.
Ihre DiGA muss in einer klinischen Studie den positiven Versorgungseffekt nachweisen, um dauerhaft im Verzeichnis gelistet zu sein. Diese sind sehr zeit- und kostenintensiv, weshalb das Budget oft nur für ein oder zwei Studien ausreicht. Daher können Sie nur eine begrenzte Anzahl an Produkt-Ansätzen ausprobieren bzw. evaluieren. Der Druck steigt, dass der erste Ansatz der Richtige sein muss. Demnach müssen Sie ausreichend planen, um die Erfolgswahrscheinlichkeit dieses Ansatzes zu erhöhen.
Die Planung der DiGA sollte natürlich auch die Ergebnisse aus etwaiger qualitativer und quantitativer User Research berücksichtigen. Sie können zu Beginn verschiedene Ansätze in Betracht ziehen. Sobald Sie sich jedoch für einen entschieden haben, ist ein Richtungswechsel deutlich schwieriger.
Abseits von der Produktkonzeption gilt es auch bei der Studie, auf Qualität zu achten. Wie Sie bestimmt wissen, müssen Sie eine vergleichende Studie durchführen.
Hier sind unter anderem die folgenden Fragen zu beantworten:
- Wie groß soll die Stichprobe sein?
- Für welche Patientengruppe (ICD-Code) wollen Sie konkret einen Nachweis erzielen?
- Wie erhalten Sie Zugang zu dieser Patientengruppe?
- Wie sieht deren aktuelle Versorgungsrealität aus?
- Welche ethischen Bedenken gibt es bei der Studie?
- Wie viele Vergleichsgruppen benötigen Sie?
- Wie wird bei der Gruppenzuweisung der Probanden vorgegangen?
- Welche Endpunkte sollen konkret gemessen werden – und wie wollen Sie diese erheben?
- Wie lange muss die DiGA genutzt werden, um einen positiven Versorgungseffekt nachzuweisen?
- Welchen Versorgungseffekt soll Ihre DiGA überhaupt erzielen?
Suchen Sie sich für die klinische Studie am besten einen zuverlässigen Partner, welcher speziell mit DiGA- und Software-Studien bereits Erfahrungen hat (gerne helfen wir Ihnen bei der Auswahl der CRO mit unserem Kontaktnetzwerk – schicken Sie uns einfach hierzu eine kurze Nachricht).
Außerdem steht nach Ende des Erprobungszeitraums die Preisverhandlung mit dem GKV-Spitzenverband an. Dort entscheidet sich, wie lukrativ Ihre Anwendung auf Dauer sein wird. Die Qualität Ihrer Studie und die Stärke des positiven Versorgungseffekts sind dabei enorm wichtige Verhandlungsargumente, die es Ihnen ermöglichen, einen dauerhaft höheren DiGA-Preis zu erzielen.
Unsere Empfehlung:
Prüfen Sie, ob es existierende wissenschaftliche Evidenz gibt, die suggeriert, dass Ihre DiGA einen positiven Versorgungseffekt nachweisen kann – und wenn ja, wie dieser aussieht. Fahren Sie erste qualitative und quantitative Untersuchungen, um zusätzliche Daten für die Produkt-Konzeption zu generieren. Planen Sie zudem das Studiendesign (ggf. mit einer CRO) wirklich gut, damit Sie keine Überraschungen während oder bei Abschluss der Studie zu erwarten haben. Je sauberer Ihr methodisches Vorgehen ist, desto größer ist die Chance, eine dauerhafte Zulassung zu bekommen und einen höheren Preis für Ihre DiGA zu verhandeln.
4. Arbeiten Sie mit den richtigen Medizinprodukt-Beratern zusammen
Eine DiGA muss vor dem Antrag als Medizinprodukt zugelassen werden. Gerade im Medizinprodukte-Bereich sollten Sie sich Partner suchen, die Sie zielführend hierbei beraten können. Schlechte Berater kosten Sie enorm viel Zeit und Geld. Gute Berater helfen Ihnen, zügig und kostenschonend auf den Markt zu gelangen.
Unpassende Berater zeichnen sich z.B. durch folgende Eigenschaften aus. Sie …
- … haben wenig Vorwissen in Bezug auf Standalone-Software. Die Berater kommen z.B. aus dem Hardware-Bereich und haben Schwierigkeiten, die Komplexität und das Mindset in der Standalone-Software-Entwicklung zu verstehen.
- … haben keine Erfahrung mit agiler Entwicklung, schon gar nicht in Bezug auf Medizinprodukte.
- … erklären Dinge unnötig kompliziert und zeigen keinen klaren Weg zur Umsetzung auf.
- … treiben die Beratungskosten künstlich in die Höhe, indem sie sich unersetzlich machen. Sie blockieren den Wissenstransfer zu Ihrem Unternehmen entweder wissentlich oder unbewusst.
Gute Berater hingegen …
- … finden pragmatische Lösungen, die kreativ und konkret umsetzbar sind. Gute Berater kennen die Regeln und wissen, wie man diese befolgt, ohne unnötig Ressourcen zu verbrennen.
- … erarbeiten Lösungen, die in Ihr Unternehmen passen, anstatt Ihnen umständliche ihre Standard-Prozeduren aufzuzwingen.
- … erklären Dinge in einfacher Sprache und ermöglichen somit Wissensvermittlung zu Ihrem Team. Gute Berater machen sich Stück für Stück selbst überflüssig, sodass Ihr Team irgendwann auf eigenen Beinen steht.
- … veranstalten nicht für jedes kleine Problem einen mehrtägigen Workshop, sondern helfen effizient beim Finden einer Lösung.
Vergessen Sie nicht, dass Sie jeden Monat, den Sie nicht als DiGA gelistet sind, kein Geld verdienen, aber dennoch Kosten haben. Daher ist gute Beratung absolut integral für den Erfolg Ihres Produkts.
Unsere Empfehlung:
Seien Sie sich der oben genannten Probleme bewusst und vorsichtig bei der Auswahl Ihrer Berater. Gerne helfen wir Ihnen hier mit unserem Netzwerk und vermitteln Ihnen vertrauenswürdige Berater für Ihre Problemstellung. Sprechen Sie uns einfach kurz über unser Kontaktformular hierzu an.
5. Nutzen Sie das Beratungsangebot des BfArM
In Bezug auf produkt-spezifische DiGA-Fragen sollten Sie mit dem BfArM Kontakt aufnehmen. Versuchen Sie natürlich zuerst, sich die Fragen selbst durch das Studieren des BfArM-Leitfadens und der DiGAV zu beantworten. Einige Fragen sind allerdings so individuell, dass Sie eine fallspezifische Einschätzung des Prüfers (dem BfArM) benötigen.
Zögern Sie also nicht, eine kostenpflichtige Beratung beim BfArM hierfür zu buchen. Wenn Sie Ihre Fragen gut vorbereitet haben, bekommen Sie so wichtige Antworten, die über den Erfolg Ihrer Listung im Verzeichnis entscheiden können.
Aber Vorsicht!
Wartezeiten für Beratungsgespräche mit dem BfArM sind teilweise enorm lang. Sie sollten ggf. damit rechnen, dass Sie mindestens 6 Monate auf einen Termin warten müssen.
Unsere Empfehlung:
Der BfArM-Leitfaden, sowie die Zusammenarbeit mit Beratern wird Ihnen den Großteil Ihrer Fragen beantworten. Für produktspezifische Schlüsselfragen sollten Sie sich jedoch an das BfArM wenden, um Überraschungen im Prüfprozess zu vermeiden.
6. Visieren Sie Risikoklasse I an
Aktuell herrscht weiterhin Unsicherheit, ob es unter MDR in Zukunft noch Software-Medizinprodukte der Risikoklasse I geben wird. Die aktuelle Situation (Stand 2026) beschreiben wir in unserem Fachartikel hier.
Kurz zusammengefasst: Es existiert eine gesetzliche Grauzone mit Interpretationsspielraum. In vielen Bundesländern ist aktuell aber weiterhin eine Zulassung unter Risikoklasse I möglich.
Bisher sind zum Beispiel fast alle unter MDR zugelassenen Anwendungen im DiGA-Verzeichnis der Risikoklasse I zugeordnet. In unserem DiGA-Verzeichnis für Hersteller können Sie sich darüber einen konkreten Überblick verschaffen.
Ob auch Ihr Medizinprodukt in die Risikoklasse I fällt, hängt von verschiedenen Faktoren ab. Lesen Sie dafür den oben erwähnten Artikel.
Generell geht mit einer Software der Risikoklasse IIa erheblicher Mehraufwand und Zusatzkosten für die Zertifizierung mit einer Benannten Stelle einher. Es kann sich lohnen, die Anwendung und vor allem die Zweckbestimmung so zu definieren, dass Ihr Produkt unter die Risikoklasse I fällt.
Uns ist natürlich vollkommen klar, dass eine entsprechende Änderung am Konzept nicht für jedes Produkt möglich ist. Da es aber insbesondere bei Produkten, die von Patienten verwendet werden, durchaus häufig funktionieren kann, lohnt es sich unter Umständen, über diesen Punkt nachzudenken.
Gerne beraten wir Sie hierzu und evaluieren, ob Risikoklasse I für Ihr Produkt klappen könnte.
Unsere Empfehlung:
Definieren Sie die Zweckbestimmung Ihrer Anwendung so, dass eine Klassifizierung nach Risikoklasse I potenziell möglich ist. Ab Risikoklasse IIa kommt erheblich mehr Aufwand auf Sie zu. Im Notfall können Sie Ihr Produkt später noch auf Risikoklasse IIa hochstufen und sich hierfür eine benannte Stelle suchen.
7. Verwenden Sie existierenden Code zur Umsetzung der ePA- und GesundheitsID-Anbindung
Die Anbindung an die elektronische Patientenakte (ePA) und die GesundheitsID kann eine erhebliche Herausforderung darstellen – insbesondere für Unternehmen, die sich erstmals mit diesen Schnittstellen auseinandersetzen. Die technischen Spezifikationen sind komplex, und die offizielle Dokumentation ist umfangreich und teils schwer verständlich.
Doch es gibt Abkürzungen: Statt sich mühsam durch die Spezifikationen zu arbeiten und eine eigene Implementierung von Grund auf zu entwickeln, lohnt es sich, auf existierenden Code und technische Beratung zurückzugreifen. Wer mit einem Partner zusammenarbeitet, der bereits Erfahrung mit der Integration hat, spart nicht nur Entwicklungsaufwand, sondern reduziert auch das Risiko von Implementierungsfehlern.
Unsere Empfehlung:
Nutzen Sie bestehende Implementierungen und das Know-how erfahrener Partner, um die ePA- und GesundheitsID-Anbindung effizient umzusetzen. So vermeiden Sie langwierige Trial-and-Error-Phasen und können sich schneller auf die eigentliche Entwicklung Ihres Produkts konzentrieren.
QuickBird Medical bietet sowohl wiederverwendbare technische Komponenten als auch technische Beratung hierfür an. Mehr Informationen finden Sie hier.
Sechs weitere DiGA-Tipps erhalten Sie in unserem Whitepaper…
Alle weiteren Tipps rund um die Zulassung und Zertifizierung finden Sie in unserem DiGA-Whitepaper. Einfach über das Formular downloaden.
Fazit
Erhöhen Sie die Erfolgswahrscheinlichkeit Ihrer DiGA
Die Entwicklung einer DiGA ist durch die regulatorischen Hürden kein Unterfangen, das unterschätzt werden sollte. Mit guter Planung und der richtigen Beratung stehen die Chancen jedoch gut, dass Sie die Zulassung in das DiGA-Verzeichnis schaffen. Wir hoffen, dass Ihnen die obigen Empfehlungen für die Zulassung ihrer DiGA helfen, unnötigen Aufwand und potenzielle Risiken zu vermeiden.
Weitere Informationen zu Themen wie dem DiGA-Zulassungsprozess, dem Nachweis des positiven Versorgungseffekts und der Preisverhandlung finden Sie auch in unserem DiGA- und Medical Software-Blog.
The post 13 Tipps für die Zulassung von DiGA in 2026 appeared first on QuickBird Medical.
]]>The post Änderungsentwurf der MDR (2025): Implikationen für Klasse I-Software appeared first on QuickBird Medical.
]]>Die EU-Kommission hat nun am 16. Dezember 2025 einen Änderungsvorschlag zur Medical Device Regulation (MDR) veröffentlicht. Unter anderem beinhaltet dieser Änderungsentwurf eine Änderung der berüchtigten Regel 11 zur Klassifizierung von Software-Medizinprodukten.
Die große Frage für Hersteller von digitalen Gesundheitsanwendungen (DiGA) und Medical Software: Welche Software-Produkte fallen unter dem neuen Entwurf in Klasse I, welche in Klasse IIa?
Eines der Ziele dieses Änderungsvorschlags zur MDR, ist laut EU-Kommission, Innovationen zu fördern. Daher liegt die Annahme nahe, dass im neuen Entwurf jetzt klar geregelt wird, dass Produkte mit niedrigem Risiko auch in Risikoklasse I fallen. Wie Sie weiter unten lesen können, ist dem vermutlich leider nicht so.
Wir haben uns den Entwurf und alle in diesem Bezug relevanten Dokumente im Detail angesehen. In diesem Artikel liefern wir Antworten zur aktuell geplanten Klassifizierung von Software-Medizinprodukten.
Inhaltsverzeichnis
- 1. Aktueller Stand: Klassifizierung von Software-Medizinprodukten
- 2. Geplanter Entwurf: Änderung der Regel 11 für Software-Medizinprodukte
- 3. Interpretationen der MDR-Änderungen zur Risikoklassifizierung
- 4. Was fällt noch in Risikoklasse I mit dem Draft für Änderungen der MDR?
- 5. Ist dieser Änderungsvorschlag final und wird er so umgesetzt?
- 6. Zeitplan: Ab wann gelten die MDR-Änderungen und die neue Regel 11?
- 7. Weitere Interpretationen
- 8. Fazit
1. Aktueller Stand: Klassifizierung von Software-Medizinprodukten
Bis die geplanten Änderungen in irgendeiner Form in der MDR landen, gelten natürlich die aktuellen Bestimmungen erst einmal weiter. In diesem Zusammenhang haben wir drei detaillierte Leitfäden geschrieben, welche Software-Medizinprodukt-Herstellern bei der Einordnung in eine MDR-Risikoklasse helfen:
- Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden
- Klasse I Software nach MDR – Geht das noch?
- MDR Risikoklasse I vs. IIa: Unterschiede für Software-Medizinprodukte
2. Geplanter Entwurf: Änderung der Regel 11 für Software-Medizinprodukte
Der wohl wichtigste Teil der MDR in Bezug auf die Klassifizierung von Software-Medizinprodukten ist die Regel 11. Diese Regel soll sich nun grundlegend ändern.
Den aktuellen Entwurf für diese Änderung finden Sie hier: Annex zum Proposal for a regulation to simplify rules on medical and in vitro diagnostic devices
Der neue Wortlaut der Regel 11 würde dann wie folgt lauten:
6.3 Rule 11
Software which is intended to generate an output that confers a clinical benefit and is used for diagnosis, treatment, prevention, monitoring, prediction, prognosis, compensation or alleviation of a disease or condition is classified as class I, unless the output is intended for a disease or condition:
- in a critical situation with a risk of causing death or an irreversible deterioration of a person’s state of health, in which case it is classified as class III;
- in a serious situation with a risk of causing a serious deterioration of a person’s state of health or a surgical intervention, or to drive clinical management in a critical situation in which cases it is classified as class IIb;
- in a non-serious situation, or to drive clinical management in a serious situation or to inform clinical management in a critical or serious situation in which cases it is classified as class IIa.’;
Gemäß der vorgeschlagenen Regel landet also jede Software zunächst in Klasse I, sofern keine höherklassifizierenden Kriterien greifen. Dies ist eine Umkehrung der bisherigen Logik und klingt aus Herstellersicht erst einmal begrüßenswert. Es klingt beinahe so, als wäre Klasse I eher die Regel als eine Ausnahme bei Software-Medizinprodukten. Bei genauerer Betrachtung lässt sich dieser Eindruck aber nicht unbedingt bestätigen.
3. Interpretationen der MDR-Änderungen zur Risikoklassifizierung
Unser Experten-Team kam bei der Analyse der vorgeschlagenen Regel 11 zu zwei möglichen Auslegungen.
Zur Interpretation der Regel verwenden wir neben dem reinen Wortlaut auch folgende Dokumente:
- MDCG 2019-11 Rev.1: Das quasi-offizielle Guidance-Dokument zur Klassifizierung und Qualifizierung von Software-Medizinprodukten unter der MDR. Hier ist insbesondere die Tabelle in Annex III relevant, in welcher auf die Risikoklassifizierung der IMDRF verwiesen wird.
- IMDRF – Software as a Medical Device: Possible Framework for Risk Categorization and Corresponding Considerations: Internationales Rahmenwerk zur risikobasierten Einordnung von SaMD. Es bewertet Software anhand der klinischen Bedeutung der erzeugten Information und der Schwere der medizinischen Situation und bildet die konzeptionelle Grundlage für die Software-Klassifizierung in der MDCG 2019-11.
Diese Dokumente helfen beim Verständnis zentraler Begriffe (z.B. „critical situation“, „drive clinical management“) des MDR-Änderungsvorschlags und beinhalten die unten verwendeten Tabellen.
3.1 Interpretation 1: Strenge Auslegung
Streng genommen kann man die Regel so lesen, dass einfach jede Software, die
- … in einer „critical situation“ verwendet wird, in Klasse III fällt.
- … in einer „serious situation“ verwendet wird, in Klasse IIb fällt.
- … in einer „non-serious situation“ verwendet wird, in Klasse IIa fällt.
Jegliche Ausnahmen (z.B. „to drive clinical management in a critical situation […] is classified as class IIb“) werden bei dieser Auslegung nicht berücksichtigt. Laut MDR gilt bezüglich der Klassifizierung nämlich immer die strengste anwendbare (Sub-)Regel. Demnach könnte man die neue Regel so auslegen, dass immer die allgemeine Regel zur Einordnung in die höhere Risikoklasse greift.
Gemäß dieser Auslegung würde man zu folgender Einstufung der Risikoklassen kommen:

Änderungsvorschlag der MDR-Regel 11 – Tabelle zur Einstufung der Risikoklassen mit strenger Auslegung
Wie Sie sehen, fehlt hier die Risikoklasse I komplett. Es gibt keine offensichtliche Argumentation, um Software in diesem Fall noch der Klasse I zuzuordnen.
Diese Auslegung scheint uns aber sehr unwahrscheinlich, da der Wortlaut der Regel 11 in diesem Fall sehr viel einfacher sein könnte und sich die EU-Kommission bestimmt etwas bei den Ausnahmen und Zusätzen gedacht hat. Rechtlich gesehen ist auch fraglich, ob man die Unterpunkte der Regel 11 als „Unterregeln“ im Sinne der MDR-Durchführungsvorschrift 3.5 bezeichnen kann (wonach quasi immer die strengste Unterregel zu Klassifizierung anzuwenden ist).
3.2 Interpretation 2: Wörtliche Auslegung
Man könnte die Regel zunächst genauso wie oben auslegen. Berücksichtigt man dann zusätzlich die explizit genannten Ausnahmen (z.B. “to drive clinical management in a critical situation […] it is classified as class IIb”), kommt man exakt zu der Tabelle in der MDCG 2019-11 Rev.1.
Gemäß dieser Auslegung würde man zu folgender Einstufung der Risikoklassen kommen:

Änderungsvorschlag der MDR-Regel 11 – Tabelle zur Einstufung der Risikoklassen mit wörtlicher Auslegung
Diese Auslegung ist sehr wahrscheinlich, denn sie würde bedeuten, dass die EU-Kommission versucht hat, die bereits veröffentlichte Tabelle im MDCG-Dokument in einen Gesetzestext zu übersetzen. Interessanterweise kam unser Team durch unsere eigene Analyse der vorgeschlagenen Regel-11-Änderung zu exakt der Tabelle im MDCG-Dokument, ohne sich an dieser zu orientieren.
Wie Sie sehen, fehlt aber auch hier die Risikoklasse I in der Zuordnung. Man müsste daher schon sehr kreativ mit der Argumentation werden, um noch Wege für Software nach Risikoklasse I zu finden.
4. Was fällt noch in Risikoklasse I mit dem Draft für Änderungen der MDR?
Die vorgeschlagene Änderung ist insbesondere für Hersteller von Produkten der Klasse I relevant, die Produkte herstellen, die nur zur Anwendung durch Patienten (medizinische Laien) bestimmt sind. Dazu gehören viele digitale Gesundheitsanwendungen (DiGA) und andere Digital Therapeutics (DTx).
Das aktuell populärste Argument dieser Hersteller zielt auf einen besonderen Wortlaut in der aktuellen MDR ab: „Software, die dazu bestimmt ist, Informationen zu liefern, die zu Entscheidungen für diagnostische oder therapeutische Zwecke herangezogen werden, gehört zur Klasse IIa, […] Sämtliche andere Software wird der Klasse I zugeordnet.“ – Hersteller argumentieren hier, dass ein Patient als medizinischer Laie keine diagnostischen oder therapeutischen Entscheidungen treffen kann. Mehr dazu lesen Sie in unserem Artikel: Klasse I Software nach MDR – Geht das noch?
In der geänderten Regel 11 gibt es diesen Wortlaut aber nicht mehr, wodurch die Rechtfertigung für Risikoklasse I für viele Hersteller nicht mehr ganz einfach werden könnte. Im Gegenteil, durch die Nutzung der Begriffe der MDCG im Gesetzestext (z.B. „Situation or patient condition“ und „Significance of information“) könnten auch deren Definitionen der IMDRF mehr Relevanz bekommen. Dort werden Laien als Nutzer des Medizinprodukts explizit in der Definition einer non-serious oder serious situation inkludiert. Somit würden auch Anwendungen für Laien mindestens in Klasse IIa fallen.
Für uns wäre es denkbar, dass sich dadurch die Diskussion um Begrifflichkeiten verschiebt zu
- Was bedeutet „Clinical Management“?
- Was ist eine Situation, die weder „critical“, „serious“, noch „non-serious“ ist?
Wenn Sie einen Prüfer in Zukunft von der Einordnung in Klasse I überzeugen möchten, müssten Sie daher entweder argumentieren, dass
- die Signifikanz der von der App gelieferten Information nicht einmal „low“ ist oder
- der Zustand des Patienten nicht einmal „non-serious“ ist.
Sie würden durch diese Argumentation quasi die existierende Tabelle um eine Spalte und Reihe erweitern:

Änderungsvorschlag der MDR-Regel 11 – Mögliche Wege in Klasse I
Es ist gut argumentierbar, dass Primärpräventionsanwendungen weiterhin in Klasse I fallen. Für jegliche therapiebegleitende App wird das Eis aber dünn und es ist nicht offensichtlich, wie hierfür noch eine Klasse-I-Argumentation erreicht werden kann.
Zumindest dann, wenn der Entwurf der MDR-Änderung wirklich so umgesetzt wird. Das ist aber alles andere als sicher und Adaptionen sind zu erwarten. Das bringt uns zu unserem nächsten Punkt:
5. Ist dieser Änderungsvorschlag final und wird er so umgesetzt?
Nein. Der vorgeschlagene Wortlaut der neuen Regel 11 ist aktuell lediglich ein Entwurf. Es handelt sich um eine frühe Version im europäischen Gesetzgebungsverfahren. Erfahrungsgemäß werden solche Entwürfe im weiteren Verlauf fachlich und politisch überarbeitet. Gerade bei komplexen Themen wie Software-Klassifizierung sind Änderungen sehr wahrscheinlich. Wir gehen daher aktuell nicht davon aus, dass die Regel 11 in dieser Form unverändert in den finalen Gesetzestext übernommen wird.
6. Zeitplan: Ab wann gelten die MDR-Änderungen und die neue Regel 11?
An dieser Stelle ist zu betonen, dass eine Änderung einer EU-Verordnung meist mehrere Jahre dauert. Daher rechnen wir frühestens im Jahr 2027 mit einer gültigen Revision der MDR (Stand Januar 2026). Auch dies ist eher optimistisch und der Prozess dauert ggf. noch länger.
Jedes Proposal für eine Änderung einer EU-Verordnung wird vor Verabschiedung einem mehrstufigen Prozess unterzogen:
- Gesetzgebungsvorschlag (Legislative Proposal): Die Europäische Kommission erarbeitet einen formellen Verordnungsentwurf und legt diesen dem Europäischen Parlament und dem Rat der EU zur Prüfung vor.
- Erste Lesung (First Reading): Das Europäische Parlament und der Rat befassen sich erstmals mit dem Vorschlag, können Änderungsanträge einbringen und versuchen, eine gemeinsame Position zu finden.
- Zweite Lesung (Second Reading): Kommt es in der ersten Lesung zu keiner Einigung, folgt eine zweite Prüfung durch Parlament und Rat, bei der weitere Änderungen möglich sind oder der Vorschlag abgelehnt werden kann.
- Vermittlungsverfahren (Conciliation): Können sich Parlament und Rat weiterhin nicht einigen, wird ein Vermittlungsausschuss eingesetzt, der innerhalb einer festen Frist einen gemeinsamen Kompromisstext ausarbeitet.
- Dritte Lesung (Third Reading): Der im Vermittlungsverfahren ausgehandelte Text wird abschließend sowohl vom Europäischen Parlament als auch vom Rat angenommen oder verworfen. Erst danach kann die Verordnung in Kraft treten.
Der gesamte Prozess wird hier im Detail beschrieben.
Wir beobachten die weiteren Entwicklungen eng und aktualisieren diesen Artikel laufend. Über relevante Änderungen informieren wir in unserem Newsletter.
7. Weitere Interpretationen
Es gibt weitere Wege, den vorgeschlagenen Wortlaut der Regel 11 zu interpretieren. Fakt ist aber, dass dieser Wortlaut vermutlich nach entsprechenden Revisionen nicht genau so in der MDR landen wird. Demnach ist eine Auflistung jeder noch so unwahrscheinlichen Interpretationsmöglichkeit zum aktuellen Zeitpunkt bislang nicht zielführend.
8. Fazit
Der Änderungsvorschlag der Regel 11 erscheint uns nach der obigen Auslegung gravierend. Die meisten Software-Medizinprodukte der Klasse I würden mit der neuen Regel sehr klar in Klasse IIa (oder höher) fallen, was für viele Hersteller ein ernst zu nehmendes Problem wäre. Ob das mit Absicht ist, zweifeln wir aktuell stark an. Das würde dem erklärten Ziel der Innovationsförderung komplett widersprechen.
Auch erscheint uns die aktuelle Formulierung der Regel 11 fachlich inkonsistent. Insbesondere fehlt eine klare und nachvollziehbare Abbildung von Software mit sehr niedrigem Risiko, wie sie beispielsweise in der von der IMDRF vorgeschlagenen 3×3-Risikomatrix vorgesehen ist. Dort ist explizit eine Kategorie für geringfügige klinische Auswirkungen vorgesehen, die sachlich einer Klasse-I-Einstufung entsprechen würde. Diese Differenzierung geht im aktuellen Entwurf verloren. Daher gehen wir davon aus, dass dies ein unbeabsichtigter Fehler ist und in den kommenden Änderungen des Entwurfs noch angepasst wird.

Kategorien für Software as a Medical Device (SaMD) von der IMDRF
Generell blicken wir weiterhin hoffnungsvoll in die Zukunft: Der aktuelle Draft existiert nur in einer ersten Version. Bis so eine Änderung einer EU-Verordnung passiert, stehen noch mehrere formale Schritte im europäischen Gesetzgebungsverfahren an.
Erfahrungsgemäß wird der Wortlaut in diesem Prozess weiter angepasst und präzisiert. Dass der aktuelle Entwurf der MDR-Änderung so final in der Verordnung landet, ist unwahrscheinlich.
Wir halten Sie hierzu in unserem Newsletter auf dem neuesten Stand.
The post Änderungsentwurf der MDR (2025): Implikationen für Klasse I-Software appeared first on QuickBird Medical.
]]>The post MDR-Guide: Klinische Bewertung von Software-Medizinprodukten appeared first on QuickBird Medical.
]]>„Da wurde doch letztens eine Ärztin in der Süddeutschen interviewt! Die hat eindeutig gesagt, dass Mindfulness-Apps bei Depressionen helfen. Darauf können wir uns doch berufen, oder?“
So einfach ist das leider nicht. Unser überspitztes Beispiel würde die Aufsichtsbehörde oder eine benannte Stelle natürlich nicht überzeugen, denn die Medical Device Regulation (MDR) verlangt für Medizinprodukte ausführliche klinische Evidenz. Selbstverständlich sind auch Software-Medizinprodukte davon nicht ausgenommen.
In diesem Artikel erfahren Sie, welche Dokumente mit welchen Inhalten Sie erstellen müssen und wie Sie um das Schreckensgespenst einer klinischen Prüfung herumkommen können.
Gut zu wissen:
In diesem Artikel (wie auch in der MDR) werden zwei Begriffe voneinander unterschieden: klinische Bewertung (clinical evaluation) und klinische Prüfung (clinical investigation). Die klinische Bewertung umfasst die gesamte Sammlung klinischer Evidenz für ein Medizinprodukt. Dazu zählt die Planung der Suche klinischer Daten, dessen Durchführung und regelmäßige Aktualisierung. Die klinische Prüfung hingegen ist eine wichtige Quelle für ebenjene klinische Evidenz – und in den meisten Fällen mit sehr viel Aufwand verbunden, da Sie hier eine eigene Studie durchführen müssen.
Fokus dieses Artikels
Der Artikel fokussiert sich auf die klinische Bewertung von Medizinprodukten nach MDR. Das Hauptaugenmerk liegt dabei auf Software-Medizinprodukten, jedoch sind die meisten Inhalte auch auf andere Medizinprodukte übertragbar. Zudem verraten wir Ihnen, wie Sie im besten Fall sogar auf eine klinische Prüfung verzichten können.
Die Informationen in diesem Artikel stammen aus der MDR, der MEDDEV 2.7/1 revision 4, sowie verschiedenen MDCG Guidance Dokumenten und werden hier zusammengefasst.
Zusätzlich werfen wir in Kapitel 5 einen Blick in die Zukunft und gehen kurz auf den neuen Norm-Entwurf ISO 18969 ein, der als kommender internationaler Standard die klinische Bewertung über den gesamten Produktlebenszyklus hinweg harmonisieren soll.
Inhalte
- 1. Ziel der klinischen Bewertung
- 2. Welche Dokumente erfordert die MDR?
- 3. Muss ich eine klinische Bewertung durchführen (auch bei Klasse I)?
- 4. Muss ich die klinische Bewertung selbst erstellen?
- 5. Ein Blick in die Zukunft: Die neue ISO 18969
- 6. Hilfreiche Quellen
- 7. Fazit
1. Ziel der klinischen Bewertung
Das fundamentale Ziel der klinischen Bewertung ist der Beweis, dass ein Medizinprodukt die vom Hersteller definierte Zweckbestimmung erfüllt. Dabei sollen die angegebenen Leistungs- und Sicherheitsanforderungen nachgewiesen und unerwünschte Nebenwirkungen ausgeschlossen werden.
Zum Teil deckt sich dieses Ziel auch mit dem Risikomanagement, bei dem potenzielle Gefahren, die vom Produkt ausgehen, analysiert werden. Wenn man so will, findet die Symbiose von Risikomanagement und klinischer Bewertung in der Nutzen-Risiko-Abwägung statt.
Eine fundierte klinische Bewertung ist notwendig, damit man die Restrisiken dem evidenzbasierten Nutzen gegenüberstellen und bewerten kann.
2. Welche Dokumente erfordert die MDR?
Wenn Sie sich mit Regulatorik von Medizinprodukten eine Zeit lang beschäftigen, ist es nichts Neues mehr, dass fast alles, was ein Hersteller macht, auch festgehalten werden muss.
Auch bei der klinischen Bewertung hat die MDR klare Vorgaben, welche Dokumente vom Hersteller erstellt und gepflegt werden müssen. Diese Vorgaben sind zum Teil jedoch sehr abstrakt und werden durch eine Handvoll Guidance-Dokumente (z. B. MDCG) ergänzt.
Wir möchten in diesem Kapitel die Inhalte der vier Dokumente erläutern, die in Ihrer technischen Dokumentation enthalten sein müssen:
- Klinischer Bewertungsplan (Clinical evaluation plan bzw. CEP)
- Bericht über die klinische Bewertung (Clinical Evaluation Report bzw. CER)
- Plan zur klinischen Nachbeobachtung nach dem Inverkehrbringen (Post-market clinical follow-up plan bzw. PMCF plan)
- Bericht über die klinische Nachbeobachtung nach dem Inverkehrbringen (Post-market clinical follow-up report bzw. PMCF report)

Erforderliche Dokumente für die technische Dokumentation gemäß MDR
2.1 Klinischer Bewertungsplan (Clinical evaluation plan)
Der klinische Bewertungsplan ist das erste Dokument, das im Zuge der klinischen Bewertung erstellt wird. Er beschreibt – wie der Name schon sagt – die geplanten Aktivitäten während der klinischen Bewertung, sowie Anleitungen zur Suche klinischer Daten und Kriterien für deren Bewertung.
Betrachten wir am besten die einzelnen Anforderungen der MDR an den klinischen Bewertungsplan, um ein klareres Bild von den Informationen zu bekommen, die enthalten sein müssen. Diese finden sich in Anhang XIV Teil A Punkt 1 Buchstabe a) der MDR. Untenstehend listen wir jede der dort genannten Anforderungen für einen klinischen Bewertungsplan auf und gehen jeweils kurz darauf ein:
„Bestimmung der grundlegenden Sicherheits- und Leistungsanforderungen, die mit relevanten klinischen Daten zu untermauern sind;“
- Werfen Sie einen Blick in den Anhang I der MDR, um die grundlegenden Sicherheits- und Leistungsanforderungen für Ihr Produkt zu identifizieren. Im Anschluss überprüfen Sie, für welche Anforderungen noch klinische Daten benötigt werden. Die grundlegenden Sicherheits- und Leistungsanforderungen beschreiben alle Anforderungen, die ein Medizinprodukt erfüllen muss. Beispiele hierfür sind Vorgaben an die chemische Zusammensetzung und das Labeling. Wie Sie schon erkennen können, ist nicht jede Anforderung für jedes Produkt zutreffend. Gerade auf reine Softwareprodukte können viele Anforderungen nicht angewandt werden. Es liegt beispielsweise auf der Hand, dass Themen wie „chemische, physikalische und biologische Eigenschaften“ oder „Schutz vor Strahlung“ für Software-Code wenig Relevanz haben. Andere Anforderungen hingegen, wie die ausreichende Genauigkeit von Messfunktionen oder die Fähigkeit zur Erfüllung der Zweckbestimmung bei der Anwendung durch Laien könnten für Softwareprodukte durch klinische Daten untermauert werden.
„Spezifizierung der Zweckbestimmung des Produkts;“
- Vermutlich haben Sie bereits eine Zweckbestimmung für Ihr Produkt geschrieben. Falls nicht, sollten Sie sich schleunigst damit befassen! Die Zweckbestimmung wird Ihnen bei der Entwicklung eines Medizinprodukts noch häufiger über den Weg laufen. Diese ist unter anderem auch der entscheidende Faktor, wenn es um die Bestimmung der Risikoklasse geht und legt fest, welchen Sinn Ihr Produkt überhaupt hat. Eine frühzeitige Definition hilft also nicht nur bei der klinischen Bewertung. Eine Anleitung zur Formulierung der Zweckbestimmung finden Sie in diesem Artikel: Formulierung der Zweckbestimmung für (Software-)Medizinprodukte
„Genaue Spezifizierung der vorgesehenen Zielgruppen mit klaren Indikationen und Kontraindikationen;“
- Dieser Punkt ist fast selbsterklärend, bedenken Sie aber bei der Spezifizierung der Zielgruppen auch verschiedene Anwendergruppen. Beispielsweise wird Ihre Software von Medizinern verwendet und nicht nur von Patienten. Zusätzlich kann es hilfreich sein, neben der Indikation auch weitere Parameter zur Eingrenzung der Zielgruppe heranzuziehen (z.B. Alter, Sprachkenntnisse, etc.). Berücksichtigen Sie auch die Liste in Appendix A3 der MEDDEV-2.7/1 revision 4, um relevante Einschlusskriterien zu identifizieren.
„Detaillierte Beschreibung des angestrebten klinischen Nutzens für die Patienten, mit relevanten konkreten Parametern für das klinische Ergebnis;“
- Die MDR definiert den klinischen Nutzen folgendermaßen: „Klinischer Nutzen“ bezeichnet die positiven Auswirkungen eines Produkts auf die Gesundheit einer Person, die anhand aussagekräftiger, messbarer und patientenrelevanter klinischer Ergebnisse einschließlich der Diagnoseergebnisse angegeben werden, oder eine positive Auswirkung auf das Patientenmanagement oder die öffentliche Gesundheit;
- Ein klinischer Nutzen könnte also sein, dass sich die Lebensqualität eines Menschen verbessert. Im zweiten Schritt stellt sich die Frage, wie dieser Nutzen gemessen werden kann. Hier müssen Sie also einerseits definieren, welchen klinischen Nutzen Ihr Produkt anstrebt und wie Sie planen, diesen Nutzen nachzuweisen. Hierzu liefert der Appendix 7.2 der MEDDEV-2.7/1 revision 4 zusätzlichen Input.
- Der klinische Nutzen beschreibt die erwartete Leistung eines Produkts, welche aus der Zweckbestimmung abgeleitet wird. Er hat die Eigenschaft, dass er klar beobachtbar sein muss. Daher sollten Sie hier konkret messbare Faktoren spezifizieren, wie z.B. „Reduktion depressiver Symptomatik“ oder „Steigerung der Lebensqualität“.
„Spezifizierung der für die Prüfung der qualitativen und quantitativen Aspekte der klinischen Sicherheit anzuwendenden Methoden unter deutlicher Bezugnahme auf die Bestimmung der Restrisiken und Nebenwirkungen;“
- Methoden, die angewandt werden, um die klinische Sicherheit des Produkts, insbesondere auch Restrisiken und Nebenwirkungen zu evaluieren. Entscheidend ist hierbei die Bewertung von gefundenen klinischen Daten. Dazu zählt unter anderem, welche Datenbanken Sie durchsuchen möchten, welche Suchfilter Sie anwenden und nach welchen Kriterien Sie gefundene Quellen bewerten. Beispielsweise sollte aus Ihrem Dokument hervorgehen, dass eine randomisierte kontrollierte Studie mehr Aussagekraft hat als ein Experteninterview. Kapitel 9.3.2. Der MEDDEV-2.7/1 revision 4 liefert hilfreiche Informationen zur Bewertung der Relevanz von Quellen. Eine spannende Liste an Bewertungskriterien für Studien finden Sie im Appendix 6 dieses Dokuments. Diese umfasst beispielsweise die Stichprobengröße und die Offenlegung der angewandten Methoden.
„Nichterschöpfende Liste und Spezifizierung der Parameter zur auf dem neuesten medizinischen Kenntnisstand beruhenden Bestimmung der Vertretbarkeit des Nutzen-Risiko-Verhältnisses für die verschiedenen Indikationen und die Zweckbestimmung bzw. Zweckbestimmungen des Produkts;“
- Hier werden die Parameter spezifiziert, um die Vertretbarkeit des Nutzen-Risiko-Verhältnisses zu bewerten. Wichtig ist es hierbei, den aktuellen medizinischen Kenntnisstand mit einzubeziehen und auch die Wirksamkeit und Risiken anderer Methoden zu berücksichtigen.
„Angabe, wie Fragen hinsichtlich des Nutzen-Risiko-Verhältnisses für bestimmte Komponenten wie die Verwendung pharmazeutischer, nicht lebensfähiger tierischer oder menschlicher Gewebe zu klären sind […];“
- Dieser Punkt kann von Software-Herstellern ignoriert werden.
„Klinischer Entwicklungsplan: von explorativen Studien, wie Studien zur Erstanwendung am Menschen („First-in-man“-Studien), Durchführbarkeitsstudien und Pilotstudien bis hin zu Bestätigungsstudien, wie pivotale klinische Prüfungen, und einer klinischen Überwachung nach dem Inverkehrbringen gemäß Teil B dieses Anhangs, unter Angabe von Etappenzielen und Beschreibung möglicher Akzeptanzkriterien;“
- Der klinische Entwicklungsplan umfasst alle Schritte, die zur Sammlung und Schaffung klinischer Daten geplant sind. Gemeint sind hierbei insbesondere wissenschaftliche Untersuchungen, die Sie zu Ihrem Produkt planen.
- Im klinischen Entwicklungsplan werden auch geplante klinische Prüfungen angegeben und auf die entsprechenden Dokumente wird verwiesen.
- Auch eine geplante Literaturrecherche wird unter diesem Punkt abgebildet.
Wenn Sie planen, die klinische Bewertung unter Bezugnahme auf ein oder mehrere gleichartige Produkte durchzuführen, können Sie in diesem Plan auch schon die Produkte listen, welche auf Gleichartigkeit untersucht werden. Die Gleichartigkeit wird im Bericht über die klinische Bewertung demonstriert.
2.2 Bericht über die klinische Bewertung (Clinical evaluation report)
Nachdem der klinische Bewertungsplan das zentrale Vorgehen bei der klinischen Bewertung beschreibt, enthält der Bericht alle daraus resultierenden Daten, die zur klinischen Bewertung herangezogen werden. Er resultiert sozusagen aus dem klinischen Bewertungsplan und enthält eine Aussage dazu, ob Ihr Produkt seine Zweckbestimmung und grundlegende Sicherheits- und Leistungsanforderungen erfüllt (einen Leitfaden zur Formulierung Ihrer Zweckbestimmung finden Sie in diesem Artikel).
Ganz konkret sollte Ihr Bericht über die klinische Bewertung folgenden Anforderungen genügen (Appendix A10, MEDDEV-2.7/1 revision 4):
- Der Bericht muss für Drittparteien verständlich sein und die klinischen Daten so zusammenfassen, dass Ihre Aussagen und Schlussfolgerungen nachvollziehbar sind.
- Die vom Hersteller erzeugten Daten (z.B. Ergebnisse einer klinischen Prüfung) müssen zusammengefasst und referenziert sein.
- Beim Vergleich mit gleichartigen Produkten:
- Die Gleichartigkeit beider Produkte muss nachvollziehbar beschrieben werden.
- Alle Unterschiede zwischen Ihrem Produkt und dem gleichartigen Produkt müssen beschrieben werden. Zusätzlich benötigen Sie eine Erklärung, warum die Produkte dennoch als gleichartig zu betrachten sind.
- Der aktuelle Stand der Technik muss vollständig zusammengefasst und mit Literatur untermauert sein. Stand der Technik? Schon wieder ein Begriff, bei dem uns die MDR mit einer eindeutigen Definition im Stich lässt. Zum Glück gibt es eine nähere Spezifikation in der ISO 14971, wonach der Stand der Technik die gegenwärtig und allgemein anerkannte gute Praxis bei Technologie und Medizin umfasst. Es handelt sich also nicht um die technologisch und wissenschaftlich fortgeschrittensten Methoden, sondern um jene Produkte, Behandlungsmethoden, etc., welche in der Praxis bereits Anwendung finden und am Markt erhältlich sind.
- Der Bericht muss eine Erklärung beinhalten, warum das Nutzen-Risiko-Verhältnis in Bezug auf den Stand der Technik akzeptabel ist.
- Falls es Ihr Produkt in mehrfacher Ausführung gibt, müssen Sie erklären, dass die klinischen Daten für alle Varianten und Nutzergruppen ausreichend sind.
- Die Konformität mit allen zutreffenden Sicherheits- und Leistungsanforderungen muss bestätigt werden.
- Der Bericht muss eine Aussage darüber enthalten, dass das von Ihnen bereitgestellte Informationsmaterial auch mit den Inhalten des Berichts übereinstimmt.
- Alle verbleibenden Risiken und unbeantworteten Fragen, müssen gelistet werden. Diese müssen dann in der Überwachung nach dem Inverkehrbringen (Post-Market Surveillance) adressiert werden.
- Der Bericht muss ein Datum haben und die Lebensläufe (oder andere Qualifikationsnachweise) der beteiligten Personen enthalten. Zudem benötigen Sie eine Interessenerklärung aller Beteiligten. Lassen Sie den finalen Bericht am besten von allen unterschreiben.
2.3 Klinische Nachbeobachtung nach dem Inverkehrbringen (Plan und Bericht)
Die klinische Bewertung vor dem Markteintritt eines Produkts ist nur eine Momentaufnahme und bildet den aktuellen Kenntnisstand ab – dieser kann sich bekanntlich ändern. Beispielsweise führen Mediziner heute keinen Aderlass mehr durch und verzichten auf den Einsatz von Zigaretten bei Kopfschmerzpatienten. Doch diese Maßnahmen zählten irgendwann einmal zum aktuellen medizinischen Kenntnisstand. Deren schwerwiegende Folgen wurden erst über die Zeit erkannt.
Als Hersteller eines Medizinprodukts müssen Sie also sicherstellen, dass neue Erkenntnisse aus der Forschung und spezifisch zu Ihrem Produkt entdeckt werden – und dafür schreibt die MDR die „Klinische Nachbeobachtung nach dem Inverkehrbringen“ vor. Im Englischen heißt diese „Post-market clinical follow-up“ und lässt sich mit PMFC abkürzen.
Die PMCF beschreibt das kontinuierliche Sammeln und Analysieren klinischer Daten, die mit Ihrem Produkt in Verbindung stehen. Auch hier sollen Sie einen Plan und einen Bericht anfertigen.
Der PMCF-Plan leitet sich aus dem klinischen Bewertungsplan und den offenen Fragen und Unklarheiten aus dem Bericht über die klinische Bewertung ab. Er beschreibt – ähnlich wie der klinische Bewertungsplan – die geplanten Aktivitäten in der PMCF-Phase und viele Inhalte können auch eins zu eins übernommen werden.
Hierzu zählt zum Beispiel:
- Literaturrecherche in wissenschaftlichen Datenbanken
- Durchsuchung von Sicherheitsdatenbanken (z.B. BfArM safety database)
- Auswertung von Nutzerfeedback
- Durchführung von Studien zum Produkt
Der PMCF-Plan definiert auch, in welchem Intervall diese Aktivitäten stattfinden. In jedem Zyklus wird dann ein PMCF-Bericht erstellt, in dem die jeweiligen Ergebnisse dokumentiert und deren Auswirkungen auf den Bericht über die klinische Bewertung beschrieben werden.
Wichtig: Relevante Erkenntnisse aus dem PMCF-Bericht werden in den Bericht über die klinische Bewertung integriert. Außenstehende sollen in diesem Bericht über die klinische Bewertung also jederzeit das gesamte Wissen zum Produkt finden können – von der initialen Bewertung bis zum letzten PMCF-Zyklus.
Für die PMCF-Dokumente stellt die MDCG wertvolle Vorlagen bereit, an denen Sie sich orientieren können: MDCG 2020-7 für den Plan und MDCG 2020-8 für den Bericht.
Unterschied zwischen PMCF und Post-Market-Surveillance
Praktisch gesehen ist die PMCF Teil der Überwachung nach dem Inverkehrbringen (Post-Market Surveillance (PMS)). Bei der PMS geht es in erster Linie darum, kontinuierlich zu überwachen, ob ein Medizinprodukt seinen gewünschten Zweck erfüllt und sicher ist. Relevante Informationen können aus verschiedenen Quellen stammen, wie etwa aus direktem User Feedback oder der Auswertung von Nutzungsdaten. Wenn Ihre PMCF ausschließlich auf Literatur basiert, kann man sie als Subset von PMS betrachten. Eine Unterscheidung der beiden Begriffe ist insbesondere dann sinnvoll, wenn im Zuge der PMCF auch klinische Prüfungen und andere Studien durchgeführt werden.
3. Muss ich eine klinische Bewertung durchführen (auch bei Klasse I)?
Ja, eine klinische Bewertung muss in jedem Fall durchgeführt werden und ist für alle Medizinprodukte – auch für Risikoklasse I – verpflichtend.
Der Aufwand, den man für die klinische Bewertung einrechnen muss, kann sich allerdings gravierend unterscheiden. Teilt man die klinische Bewertung in einzelne Arbeitspakete auf, merkt man schnell, dass es einen besonders großen Block gibt – die klinische Prüfung.
MDR-Definition: „Klinische Prüfung“ bezeichnet eine systematische Untersuchung, bei der ein oder mehrere menschliche Prüfungsteilnehmer einbezogen sind und die zwecks Bewertung der Sicherheit oder Leistung eines Produkts durchgeführt wird;
Konkret handelt es sich bei der klinischen Prüfung um eine wissenschaftliche Untersuchung, die beim BfArM angemeldet werden muss. Es ist klar, dass dies oft mit enormen Kosten und erheblichem Zeitaufwand verbunden ist. Das Ganze kann aber unter Umständen sogar vermieden werden. Wenn es ein sogenanntes gleichartiges Medizinprodukt gibt, welches Ihrem Produkt derart ähnlich ist, dass Sie sich auch auf die Daten dieses Produkts berufen können, kommt man um die klinische Prüfung herum. Konkret kann dies eine Zeitersparnis von mehreren Monaten bedeuten – ganz zu schweigen von den Kosten.
3.1 Wie man gleichartige Produkte unter der MDR findet
Vermutlich geht es Ihnen nun, wie den meisten Herstellern und Sie stellen sich die Frage, ob es in Ihrem Fall ein geeignetes Produkt gibt, das bereits als Medizinprodukt am Markt ist und zu dessen klinischen Daten Sie Zugriff haben. Doch wie finden Sie heraus, ob eine Gleichartigkeit besteht?
Dafür benennt die MDR konkrete Kriterien zum Abgleich der Gleichartigkeit zweier Produkte:
Technisch: Das Produkt ist von ähnlicher Auslegung, wird unter ähnlichen Anwendungsbedingungen angewandt, hat ähnliche Spezifikationen und Eigenschaften einschließlich […] Software-Algorithmen, verwendet gegebenenfalls ähnliche Bereitstellungsmethoden und hat ähnliche Funktionsgrundsätze und entscheidende Leistungsanforderungen.
Biologisch: Ist für Software irrelevant.
Klinisch: Das Produkt wird unter der gleichen klinischen Bedingung oder zum gleichen klinischen Zweck, einschließlich eines ähnlichen Schweregrads und Stadiums der Krankheit, an der gleichen Körperstelle und bei ähnlichen Patientenpopulationen in Bezug auf u.a. Alter, Anatomie und Physiologie angewandt, hat die gleichen Anwender und erbringt eine ähnliche, maßgebliche und entscheidende Leistung im Hinblick auf die erwartete klinische Wirkung für eine spezielle Zweckbestimmung.

Kriterien für die Gleichartigkeit gemäß MDR
Für Software-Hersteller bleiben also zwei Punkte übrig: klinische und technische Gleichartigkeit. Die klinische Gleichartigkeit ist vergleichsweise einfach zu überprüfen. Ein Blick in die Zweckbestimmung oder Bedienungsanleitung anderer Anwendungen sollte hier bereits einen guten Aufschluss geben. Je nachdem, in welchem Entwicklungsstadium das eigene Produkt ist, können Hersteller hier auch z.B. noch Anpassungen bei ihrem Produkt oder der angestrebten Zielgruppe vornehmen, um Gleichartigkeit zu gewährleisten. Gerade bei einem initialen Markteintritt kann dies durchaus sinnvoll sein.
Bei der Prüfung der technischen Gleichartigkeit wird es allerdings schon schwieriger. Einer der entscheidenden Punkte ist hierbei die Bewertung der Software-Algorithmen. Aber bis wohin wird ein Algorithmus als „ähnlich“ definiert? Müssen zwei Algorithmen den exakt gleichen Softwarecode haben, um gleichartig zu sein? Ist die Auswertung zweier Fragebögen ähnlich, wenn bei beiden ein Summen-Score berechnet wird, selbst wenn sich die Fragen inhaltlich voneinander unterscheiden? Ist der Algorithmus zur Steuerung eines Blutdruckmessgeräts ähnlich wie jener zur Steuerung des Geräts von einem anderen Hersteller?
Fragen über Fragen, die auf Basis der verfügbaren Informationen nur schwer zu beantworten sind. Zumindest gibt es hierzu eine genauere Ausführung in der MDCG 2020-5, welche auch eine hilfreiche Tabelle in Anhang I beinhaltet. Diese besagt, dass es vorrangig um die Funktionsgrundsätze und die Zweckbestimmung geht, wenn man Software-Algorithmen miteinander vergleicht. Glücklicherweise ist es laut MDCG nicht notwendig, eine Äquivalenz des Softwarecodes zu beweisen.
Zitat aus der MDCG 2020-5: „It is the functional principle of the software algorithm, as well as the clinical performance(s) and intended purpose(s) of the software algorithm, that shall be considered when demonstrating the equivalence of a software algorithm. It is not reasonable to demand that equivalence is demonstrated for the software code, provided it has been developed in line with international standards for safe design and validation of medical device software.“ – Achtung: Die MDCG-Dokumente und deren Interpretationen der MDR sind rechtlich nicht bindend. Jedoch gelten sie als die wohl valideste Quelle zur Umsetzung der MDR-Anforderungen.
Ein wichtiger Hinweis an dieser Stelle: Wenn Sie sich auf ein gleichartiges Produkt beziehen, müssen Sie auch einen geeigneten Zugang zu den dazugehörigen klinischen Daten haben. Dabei durchsuchen Sie am besten zuerst wissenschaftliche Datenbanken nach relevanten Veröffentlichungen, oder Sie wenden sich direkt an den Hersteller. Eine relevante Anlaufstelle ist das Deutsche Register Klinischer Studien (DRKS). Das DRKS ist das zentrale Register für klinische Studien in Deutschland und Sie finden dort auch Untersuchungen zu Medizinprodukten.
3.2 Wenn man kein gleichartiges Produkt findet
Sollten Sie kein gleichartiges Produkt finden, gibt es für Sie leider nur eine Antwort: Sie müssen in den sauren Apfel beißen und eine klinische Prüfung durchführen. Es gibt eine Ausnahme, wenn Sie auf Basis von MDR, Artikel 61, Satz (10) begründen können, dass ein Nachweis auf Basis klinischer Daten für Ihr Produkt ungeeignet ist.
4. Muss ich die klinische Bewertung selbst erstellen?
Grundsätzlich ist die klinische Bewertung Teil der technischen Dokumentation Ihres Produkts, welche Sie als Hersteller erstellen und laufend pflegen müssen. Auch wenn Sie die Erstellung auch auslagern können, ist sie in den meisten Fällen jedoch auch von Ihnen selbst durchführbar – auch wenn Sie nur begrenzte Vorkenntnisse haben.
Selbst Start-ups haben klinische Experten (Mediziner oder andere Menschen mit wissenschaftlichem Hintergrund) oft bereits an Bord, welche ein grundlegendes Verständnis von klinischen Studien haben. Sie profitieren von der Durchführung einer klinischen Bewertung insofern, als Sie und Ihr Team durch das Lesen klinischer Studien Ihren Markt und Ihre Zielgruppe besser verstehen. Sie erhalten einen wissenschaftlichen Einblick in wirksame und möglicherweise wirkungslose Methoden und wissen auch nach dem Inverkehrbringen genau, worauf sie achten müssen und welche Daten in Zukunft relevant sind.
Bei der Durchführung einer klinischen Bewertung ist ein gewisses wissenschaftliches Verständnis wichtig, um die gefundenen Quellen einordnen und bewerten zu können. Wenn Ihnen Begriffe wie „randomisierte kontrollierte Studien“, „Stichprobengröße“ und „statistische Signifikanz“ nicht völlig fremd sind, haben Sie in den meisten Fällen gute Chancen, Ihre klinische Bewertung innerhalb weniger Wochen selbst zu erstellen (ausgenommen einer klinischen Prüfung).
Wir raten Ihnen auf jeden Fall dazu, sich zumindest grundlegend mit der Thematik auseinanderzusetzen – zu Beginn wirkt es oft komplizierter, als es eigentlich ist. Am Ende können Sie sich immer noch dazu entscheiden, das Thema auszulagern.
Für die Durchführung einer klinischen Prüfung lohnt es sich hingegen oft, einen entsprechenden Anbieter zu beauftragen. Diese haben bessere Möglichkeiten zur Rekrutierung von Studienteilnehmern und sind mit dem Design und der Durchführung solcher Untersuchungen vertraut. Wer hier keine tiefgreifende Erfahrung mitbringt, läuft Gefahr, Fehler zu machen, welche gravierende Auswirkungen haben können. Wird eine Studie beispielsweise falsch designt, könnte am Ende monatelange Arbeit umsonst gewesen sein. Geeignete Anbieter sind zum Beispiel Clinical Research Organizations (CRO), welche in den meisten Fällen über einen breiten Expertenpool im wissenschaftlichen, statistischen und medizinischen Bereich verfügen. Doch auch Hochschulen und Universitäten sind als Partner denkbar.
Achtung: Bei der Auswahl eines passenden Partners sollten Sie darauf achten, dass die durchgeführte Untersuchung auch den Anforderungen an klinische Prüfungen der MDR und des Medizinprodukterecht-Durchführungsgesetzes (nur für Deutschland relevant) genügt. Das trifft auf die meisten CRO zu, Universitäten sind hingegen in erster Linie am Erkenntnisgewinn und an Publikationen interessiert und mit harmonisierten Normen und Gesetzen nicht vertraut. Besprechen Sie Ihr Vorhaben also bereits vorab im Detail, damit es hier nicht zu Missverständnissen kommt.
Natürlich kann auch die gesamte klinische Bewertung ausgelagert werden. Das ist besonders dann hilfreich, wenn Sie selbst keine Erfahrungen mit klinischen Untersuchungen und den regulatorischen Anforderungen haben – oder wenn Sie es sich einfach leisten können und Angst davor haben, Fehler zu machen.
Ein Vorteil davon ist, dass Sie die Unabhängigkeit der involvierten Personen recht plausibel belegen können. Das kann bei der Rechtfertigung der klinischen Nachweise nach außen helfen. Vorausgesetzt, niemand erhält einen Bonus, wenn die gefundenen Daten möglichst gut zu den Aussagen des Herstellers passen. Einem Gründer eines Health-Start-ups könnte man hingegen schon eher unterstellen, dass er sein Produkt in einem guten Licht dastehen lassen will und nur ausgewählte Fakten in seinen Bericht mit aufnimmt.
Die Nachteile hingegen sind die Kosten und das Know-how, das Ihrem Team dadurch eventuell verloren geht, wenn die Suche und Analyse klinischer Studien von Externen übernommen wird.
5. Zukunft: Die neue ISO 18969
Während die MEDDEV 2.7/1 Revision 4 aktuell noch der unangefochtene Goldstandard bleibt, wirft ein neuer internationaler Standard bereits seine Schatten voraus: die ISO 18969 „Clinical evaluation of medical devices“. Der Entwurf wurde im Jahr 2025 veröffentlicht und befindet sich nun in der öffentlichen Kommentierungsphase.
Die ISO 18969 beschreibt auf internationaler Ebene, wie klinische Bewertungen systematisch über den gesamten Produktlebenszyklus hinweg durchgeführt werden sollen. Sie gibt unter anderem präziser vor, welche Evidenzquellen herangezogen werden dürfen und wie die klinische Bewertung in das Qualitätsmanagement eingebettet wird.
Unsere Empfehlung: Bis die Norm final verabschiedet ist, orientieren Sie sich weiterhin an der MDR und der MEDDEV-Leitlinie. Dennoch lohnt sich ggf. ein Blick in den Entwurf der ISO 18969, um frühzeitig ein Gefühl für die mögliche kommende Harmonisierung zu bekommen und Ihren Prozess zukunftssicher aufzustellen.
6. Hilfreiche Quellen
MDR: Zentrales Werk, aus dem die Notwendigkeit und alle Anforderungen an die klinische Bewertung abgeleitet werden.
MEDDEV-2.7/1 revision 4: Leitlinie zur Erstellung der klinischen Bewertung. Zwar wurde das Dokument nicht explizit für die MDR verfasst, jedoch sind die meisten Inhalte nach wie vor aktuell.
ISO 18969: Entwurf des kommenden internationalen Standards zur klinischen Bewertung von Medizinprodukten. Er beschreibt die systematische Durchführung über den gesamten Produktlebenszyklus.
MDCG 2020-5: Leitlinie zur Evaluierung der Gleichartigkeit zweier Produkte
MDCG 2020-7: Vorlage für einen PMCF-Plan
MDCG 2020-8: Vorlage für einen PMCF-Bericht
Formulierung der Zweckbestimmung für (Software-)Medizinprodukte: Leitfaden zur Erstellung der Zweckbestimmung für Ihr Produkt
Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden: Anleitung zum Finden der Risikoklasse Ihres Produkts nach MDR
Leitfaden: Ist Ihre App ein Medizinprodukt?: Hier erfahren Sie, ob es sich bei Ihrem Produkt um ein Medizinprodukt nach MDR handelt.
7. Fazit
Der Aufwand, der mit der klinischen Bewertung einhergeht, hängt enorm von der Notwendigkeit einer klinischen Prüfung und dem Umfang der Literaturrecherche ab. Zwar ist es unter der MDR möglich, sich auf gleichartige Produkte zu berufen, die bereits am Markt sind, jedoch bedarf diese Gleichartigkeit einer zufriedenstellenden Erklärung. Gerade für Start-ups ist es sehr sinnvoll, den Markt intensiv zu durchforsten, um derartige Produkte ausfindig zu machen. Schließlich sind klinische Prüfungen teuer und dauern mehrere Monate bis Jahre. Die Chancen, ein geeignetes Produkt zu finden, sind natürlich von Fall zu Fall unterschiedlich und beim Zugang zu den passenden klinischen Daten ist man im Zweifelsfall auf die Gunst des Herstellers angewiesen.
Auch wenn sich die klinische Bewertung erst mal sehr komplex anhört und nach viel Aufwand klingt, sollten sich auch kleinere Unternehmen und Start-ups nicht gleich abschrecken lassen. In vielen Fällen kann sie intern durchgeführt werden und bedarf keiner externen Dienstleister. Dabei können Unternehmen stark profitieren, da sie neues Wissen generieren und der Aufwand für klinische Bewertungen in Zukunft besser abgeschätzt werden kann. Falls eine klinische Prüfung notwendig ist, greifen Unternehmen oft auf „Clinical Research Organizations“ (CROs) zurück.
Falls Sie eine Medical Software oder eine DiGA planen und Unterstützung bei der Entwicklung benötigen, setzen Sie sich gerne mit uns in Kontakt und wir besprechen das Ganze zusammen unverbindlich. Wir entwickeln Medical Apps und Medical Software für Unternehmen auf Auftragsbasis.
Weitere Fragen? Rufen Sie uns gerne an (+49 (0) 89 54998380) oder schreiben uns eine Email ([email protected]).
The post MDR-Guide: Klinische Bewertung von Software-Medizinprodukten appeared first on QuickBird Medical.
]]>The post Klasse I Software nach MDR – Geht das noch? (Dezember 2025) appeared first on QuickBird Medical.
]]>Da in letzter Zeit immer mehr Gerüchte kursieren, dass eine Zulassung von Software-Medizinprodukten unter Risikoklasse I nicht mehr möglich ist, möchten wir mit diesem Artikel etwas Licht ins Dunkel bringen.
Dieser Artikel richtet sich vor allem an Hersteller von Medizinprodukt-Software, von denen keine oder nur geringe Risiken ausgehen. Das umfasst beispielsweise Trainingsapps, die von Patienten autonom genutzt werden und die keine Schäden am Patienten verursachen können.
Update – Juni 2025: Das Guidance-Dokument „MDCG 2019-11: Guidance on Qualification and Classification of Software […]“ liegt seit Juni 2025 in der überarbeiteten Revision 1 vor. In diesem Artikel sind sämtliche Anpassungen berücksichtigt. Eine detaillierte Übersicht der konkreten Änderungen finden Sie auch hier.
Update – Dezember 2025: Die EU-Kommission hat im Dezember 2025 einen Änderungsentwurf zur MDR veröffentlicht, der unter anderem eine Anpassung der Regel 11 zur Klassifizierung von Software-Medizinprodukten vorsieht. Auf die möglichen Auswirkungen für Risikoklasse-I-Software gehen wir im Kapitel 3 dieses Artikels ein.
Inhaltsverzeichnis
- 1. Risikoklassen der MDR
- 2. Update der offiziellen MDCG-Guidance (Revision 1) – Juni 2025
- 3. Änderungsentwurf der MDR & Implikationen für Klasse I-Software – Dezember 2025
- 4. Umgang mit Unsicherheit bei der Risikoklasse
- 5. Warum wir Risikoklasse I in der EU dringend brauchen
- 6. Fazit
1. Risikoklassen für Medizinprodukte der MDR
Da wir die Risikoklassen der MDR bereits in unserem Software-Klassifizierungs-Guide umfassend beschrieben haben, möchten wir es an dieser Stelle kurz halten:
- Die MDR unterscheidet bei Software im Kern die Risikoklassen I, IIa, IIb und III
- Entscheidend für die Bestimmung der Risikoklasse eines Produkts ist seine Zweckbestimmung
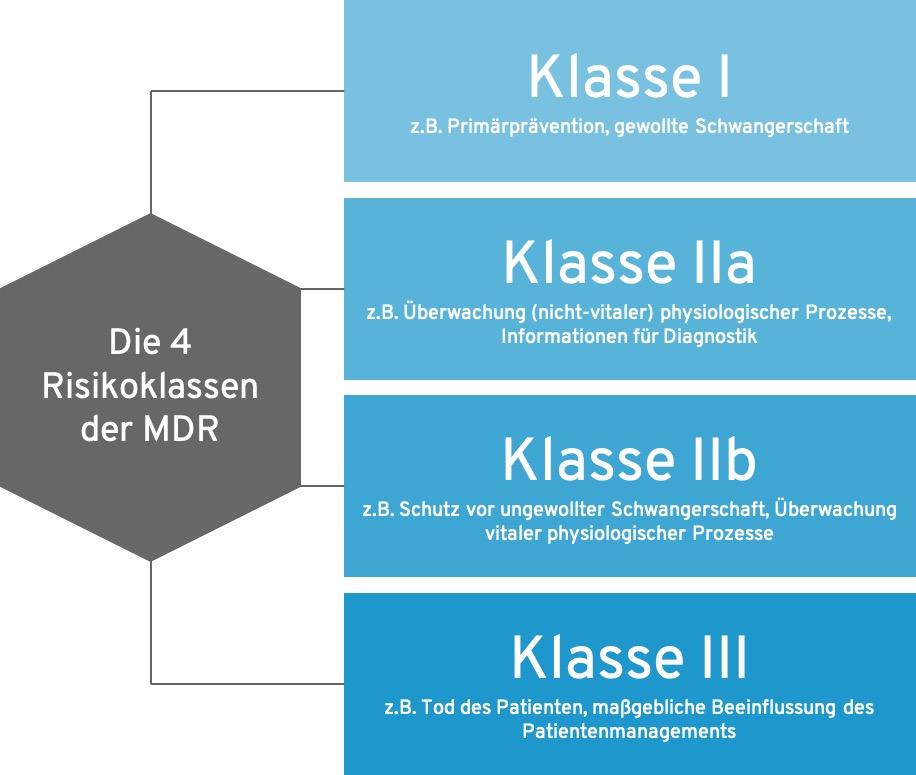
Die 4 MDR-Risikoklassen im Überblick
Falls Sie mehr über die Risikoklassifizierung von Software-Medizinprodukten erfahren möchten, helfen Ihnen folgende Leitfäden von uns:
- Qualifizierung von Software-Medizinprodukten – Leitfaden: Ist Ihre App ein Medizinprodukt?
- Klassifizierung von Software-Medizinprodukten – Leitfaden: In welche Risikoklasse fallen Sie?
Die Zulassung von Produkten der Risikoklasse I ist im Vergleich zu höherklassigen Produkten sehr viel ressourcenschonender und schneller möglich. Daher stellen sich viele Hersteller die Frage, ob es sich bei ihrem Produkt um ein Produkt der Klasse I oder IIa handelt.
Warum das so ist, erfahren Sie im nächsten Kapitel.
1.1 Risikoklasse I oder IIa – Warum ist das wichtig?
Warum ist die Unterscheidung zwischen Klasse I und IIa überhaupt so wichtig? Und warum wird so viel darüber diskutiert?
Produkte der Risikoklasse I unterscheiden sich maßgeblich von höherklassigen Produkten.
Klasse I-Produkte …
- benötigen bei Marktzulassung kein Zertifikat einer benannten Stelle,
- können ohne Verzögerung (verursacht durch einen Audit) auf den Markt gehen und müssen keine Kosten für eine benannte Stelle tragen,
- werden lediglich unangekündigten Prüfungen durch die jeweilige Aufsichtsbehörde des Bundeslands unterzogen. Diese sind deutlich weniger aufwendig als die Zertifizierung bei einer benannten Stelle (Risikoklasse IIa und höher) und nicht kostenpflichtig.
Um das Ganze noch besser zu veranschaulichen, möchten wir hier einige Erfahrungswerte aufzeigen. Ein Klasse IIa Produkt bedeutet im Vergleich zu Klasse I:
- Verzögerung der Marktzulassung um 6–18 Monate (Dauer des Konformitätsbewertungsverfahrens einer benannten Stelle und Ausstellung des CE-Zertifikats)
- Kosten bis zu 6-stelligen Eurobeträgen für das Konformitätsbewertungsverfahren und die Zertifizierung des QMS
- Pflicht der Anzeige signifikanter Änderungen und weitere Prüfungen der benannten Stelle bei Produktänderungen nach der Zulassung
Wie lange es bis zum erfolgreichen Abschluss eines Konformitätsbewertungsverfahrens dauert, ist höchst individuell und hängt von verschiedenen Faktoren ab. In jedem Fall handelt es sich dabei um eine weitere Unbekannte, die man in der Planung seines Produkts nur schwer abbilden kann.
Die Unterscheidung zwischen Risikoklasse I und IIa entscheidet daher in vielen Fällen wirklich über die Existenz von Produkten und ganzen Unternehmen.
Deshalb ist Klasse I vor allem für Start-ups interessant, die auf eine schnelle Marktzulassung angewiesen sind und nur begrenzte Ressourcen haben.
Was genau bei einem Risikoklasse IIa-Produkt im Vergleich zu Klasse I auf Sie zukommt, erfahren Sie in einem gesonderten Fachartikel von uns: MDR Risikoklasse I vs. IIa: Unterschiede für Software-Medizinprodukte
1.2 MDR – Regel 11 – Klare Grenze zwischen Klasse I und IIa?
Die Antwort, ob Ihre Software oder App in die Risikoklasse I fallen kann, findet sich in der Medical Device Regulation (MDR) – Regel 11.
Zitat der MDR 6.3. Regel 11 (verkürzt):
Software, die dazu bestimmt ist, Informationen zu liefern, die zu Entscheidungen für diagnostische oder therapeutische Zwecke herangezogen werden, gehört zur Klasse IIa,
[…]
Sämtliche andere Software wird der Klasse I zugeordnet.
In diesem Bezug fragen sich viele Hersteller:
-
- Was sind Informationen, die zu Entscheidungen für diagnostische und therapeutische Zwecke herangezogen werden?
- Wer kann solche Informationen überhaupt “heranziehen”, um diagnostische oder therapeutische Entscheidungen zu treffen?
- Kann eine App, die einem Patienten nur Trainingsmodule und Wissen vermittelt, überhaupt die Therapie oder Diagnostik beeinflussen?
- Ist die Empfehlung, eine bestimmte Übung zu machen, schon eine Therapieentscheidung?
Überraschung: Es gibt verschiedene Möglichkeiten, diese Regel 11 zu interpretieren. Denn das BfArM, benannte Stellen und Hersteller sind sich hier nicht einig. Es existiert viel Interpretationsspielraum, weshalb es immer wieder zu Diskussionen und Gerüchten kommt. Daher möchten wir hier die 2 wichtigsten Interpretationen dieser Regel 11 beschreiben.
1.2.1 Interpretation 1: Jede Software liefert Informationen, die die Therapie oder Diagnostik beeinflussen
Nach dieser Interpretation liefert auch eine App für Patienten Informationen für Entscheidungen zur Therapie oder Diagnose von Krankheiten. Der Patient würde sich ja selbst therapieren bzw. diagnostizieren und somit greife Satz 1 aus Regel 11.
Mit dieser Interpretation gibt es streng genommen keine Klasse I Standalone-Software, da Software definitionsgemäß immer einen Output (Informationen) auf Basis eines Inputs erzeugt. Eine Ausnahme wäre beispielsweise eine App, die jedem Nutzer den exakt gleichen Inhalt anzeigt, ohne irgendeine Individualisierung. Doch hier stellt sich die Frage, ob es sich dann überhaupt um ein Medizinprodukt handelt (siehe auch unser Leitfaden „Ist Ihre App ein Medizinprodukt?“).
1.2.2 Interpretation 2: Nur medizinisches Fachpersonal kann therapeutische oder diagnostische Entscheidungen treffen
Das ist die herstellerfreundliche Interpretation der Regel 11. Demnach würde eine Trainingsapp, die lediglich von Patienten verwendet wird, in Risikoklasse I fallen. Die Begründung: Ein Laie ist nicht dazu in der Lage, therapeutische oder diagnostische Entscheidungen zu treffen. Satz 1 aus Regel 11 greift daher nicht – Informationen werden nicht für Entscheidungen für diagnostische oder therapeutische Zwecke herangezogen.
Hinweis: Anders sieht es aber bei Produkten aus, die auch medizinisches Fachpersonal in die Nutzung einbeziehen und unterstützende Informationen liefern (z. B. über ein Web Dashboard). Dies wurde beispielsweise vom Hanseatische Oberlandesgericht im Fall von Dermanostic entschieden. In diesem Fall hat ein Mitbewerber ein Gerichtsverfahren gegen Dermanostic eingeleitet. Das Gericht entschied dann, dass es sich bei Dermanostic um ein Produkt der Klasse IIa handelt und nicht um eines der Klasse I. Mehr dazu lesen Sie hier.
1.2.3 Wessen Interpretation zählt nun?
Wir sehen, dass verschiedene Stakeholder unterschiedliche Auffassungen vertreten:
-
- BfArM
- Landesaufsichtsbehörde
- Benannte Stelle
- Hersteller
- Gericht
Wenn Sie sich dazu entscheiden, den Weg zum Risikoklasse I Produkt zu gehen, ist vor allem Ihre eigene Meinung (Hersteller), aber natürlich auch die Meinung Ihrer Landesaufsichtsbehörde relevant. Diese Behörde ist nämlich in erster Linie verantwortlich für die Überwachung der Hersteller in Ihrer Region.
Auch die Entscheidung von Gerichten zählt natürlich, sofern es zu einem Rechtstreit (z.B. zwischen Ihnen und einem Konkurrenten) kommt – siehe Dermanostic .
Die Einschätzung benannter Stellen ist vor allem dann wichtig, wenn Sie Risikoklasse IIa oder höher anstreben. Bei Produkten der Risikoklasse I sind diese nämlich gar nicht eingebunden.
Das BfArM hat vor allem indirekten Einfluss auf die Risikoklassifizierung. Da es sich dabei um die Bundesbehörde handelt, kann das BfArM auch Einfluss auf die Landesaufsichtsbehörden ausüben und z.B. Empfehlungen herausgeben, denen Landesbehörden ggf. folgen.
1.3 Schlussfolgerung – geht Klasse I noch?
Ja, eine Zulassung von Software-Medizinprodukten der Risikoklasse I ist noch möglich. Allerdings ist das stark von Ihrer Landesaufsichtsbehörde abhängig. Unter Umständen kann es sich lohnen, den Sitz Ihres Unternehmens in Bundesländer zu verlagern, deren Aufsichtsbehörden „Klasse I-freundlich“ sind.
Alternativ gibt es ein weiteres spannendes Modell, das wir anbieten. Sie können uns als externen Inverkehrbringer beauftragen, Ihr Medizinprodukt auf den Markt zu bringen. Wir bieten diesen Service für Software-basierte Medizinprodukte und DiGA an: Mehr Informationen
Sprechen Sie uns gerne an, wenn Sie einen externen Inverkehrbringer für Ihre App oder andere Standalone-Software benötigen.
2. Update der offiziellen MDCG-Guidance (Revision 1) – Juni 2025
Die offizielle MDCG-Guidance zur Qualifizierung und Klassifizierung von Medical Device Software (MDSW) wurde im Juni 2025 in Revision 1 aktualisiert. Das Update wurde lange erwartet und erzeugte große Unsicherheit in der Digital Health-Szene. Die zentrale Frage im Raum: Bedeutet die neue Version das Ende von Risikoklasse I-Software?
Das ist glücklicherweise nicht der Fall. Es hat sich tatsächlich wenig in Bezug auf die (teilweise unkonkreten) Empfehlungen zur Risikoklasse I vs. IIa getan. Wir haben hier eine detaillierte Übersicht zu allen Änderungen erstellt: Alle Änderungen der MDCG 2019-11 Rev.1
3. Änderungsentwurf der MDR & Implikationen für Klasse I-Software – Dezember 2025
Am 16. Dezember 2025 hat die EU-Kommission einen Änderungsentwurf zur Medical Device Regulation (MDR) veröffentlicht. Ein zentraler Bestandteil dieses Entwurfs ist eine grundlegende Überarbeitung der Regel 11, welche wir in der aktuell gültigen Fassung oben erklärt haben.
Auf den ersten Blick wirkt der neue Wortlaut für Hersteller vielversprechend: Die Logik der Klassifizierung wird scheinbar umgedreht, sodass Software zunächst in Klasse I fällt, sofern keine höherklassifizierenden Kriterien greifen. Bei genauerer Analyse zeigt sich jedoch, dass diese Hoffnung trügerisch sein könnte. Je nach Auslegung würde der Entwurf dazu führen, dass ein Großteil medizinischer Software mindestens in Klasse IIa eingeordnet werden würde.
Wir haben den Änderungsentwurf, die neue Regel 11 sowie deren mögliche Interpretationen detailliert analysiert und die konkreten Auswirkungen auf Klasse-I-Software in einem separaten Fachartikel aufgearbeitet: Zum Fachartikel
Wichtig ist dabei: Der veröffentlichte Text ist ein früher Entwurf im europäischen Gesetzgebungsverfahren. Erfahrungsgemäß wird der Wortlaut in den kommenden Jahren weiter angepasst. Es ist daher unwahrscheinlich, dass dieser Entwurf für die Regel 11 unverändert in den finalen MDR-Text übernommen wird.
4. Umgang mit Unsicherheit bei der Risikoklasse
Wie kann ich sicher sein, dass meine Risikoklasse richtig ist? Eine absolute Rechtssicherheit gibt es bei diesem Thema eigentlich nie. Grundsätzlich ist eine höhere Klassifizierung immer “sicherer”, jedoch auch sehr viel aufwendiger und teurer.
Da sich ein solches Verfahren aber viele Firmen nicht leisten wollen oder können, möchten wir hier ein paar Möglichkeiten aufzeigen, die Sie als Hersteller eines Klasse-I-Produkts haben.
- Lesen Sie die MDCG Guidance Dokumente (v.a. MDCG 2019-11).
- Sehen Sie sich ähnliche Produkte auf dem Markt an, die unter MDR zugelassen sind. Dabei sollten Sie vor allem Produkte von Herstellern berücksichtigen, für die Ihre auch Ihre Aufsichtsbehörde verantwortlich ist.
- Holen Sie Erfahrungswerte von Experten ein. Es gibt viele Berater, die in die Zulassung vieler Klasse I-Medizinprodukte involviert waren. Diese Erfahrungen sind in vielen Fällen auf ihr Produkt übertragbar. Sprechen Sie uns gerne an, wenn Sie dabei Unterstützung benötigen.
- Holen Sie ein juristisches Gutachten ein. Ein solches wird von einem Anwalt ausgestellt und kann Ihre Argumentation vor einer Aufsichtsbehörde, aber auch vor Gericht untermauern. Es schafft zusätzliche Überzeugungskraft. Beachten Sie aber, dass ein solches Gutachten keine absolute Rechtssicherheit geben kann. Sprechen Sie uns an, wenn Sie an einem Gutachten interessiert sind. Wir vermitteln Sie gerne an einen Anwalt, der sich mit der Thematik auskennt.
4.1 Was passiert, wenn ich mein Produkt unter Risikoklasse I anmelde, obwohl ich mir unsicher bin?
Grundsätzlich können Sie das natürlich tun. Es liegt in der Verantwortung des Herstellers, die Klassifizierung seiner Produkte ordnungsgemäß durchzuführen.
ABER seien Sie sich bewusst, dass dies ein unternehmerisches Risiko darstellen kann. Einige dieser Risiken sind:
| Szenario | Gefahr | Schaden |
| Das Produkt birgt wirklich Risiken in der Anwendung | Ein Patient könnte sich verletzen | Rechtliche Konsequenzen, Reputationsschaden |
| Ein Mitbewerber leitet Gerichtsverfahren gegen Sie ein | Das Gericht entscheidet, dass Sie Ihr Produkt höher klassifizieren müssen | Produkt muss vom Markt genommen oder höher klassifiziert werden |
| Eine Aufsichtsbehörde prüft Ihr Produkt und widerspricht der Risikoklassifizierung | Die Aufsichtsbehörde ist anderer Meinung in Bezug auf die Risikoklasse Ihres Produkts | Produkt muss vom Markt genommen oder höher klassifiziert werden |
5. Warum wir Risikoklasse I in der EU dringend brauchen
Es gibt unzählige innovative Software-Produkte mit medizinischem Anwendungsfall, von denen kaum Risiken für Patienten ausgehen (z.B. Trainingsapps zur Reduktion von Ängsten). Der Zugang zu solchen Produkten wurde in den letzten Jahren (z.B. durch die Einführung von DiGA) enorm gefördert und tausende Patienten nutzen solche Produkte täglich effektiv zur Verbesserung ihres Gesundheitszustands. Das entlastet nicht nur Patienten, sondern das gesamte Gesundheitssystem.
Der Aufwand für eine Zulassung unter Risikoklasse IIa steht aus unserer Sicht in keinem Verhältnis zu den meist sehr geringen Risiken, die von Patienten-fokussierten Laienprodukten ausgehen.
Solche Produkte kommen zudem oft aus dem Startup-Umfeld, welches meist mit limitierten Ressourcen auskommen muss. Durch eine Überregulierung (und hart gesagt – das Unmöglich-Machen von Klasse I Software) erstickt man solche Innovationen im Keim. Kleine Unternehmen können es sich schlichtweg nicht leisten, ein Konformitätsbewertungsverfahren mit einer benannten Stelle zu durchlaufen – weder zeitlich, noch finanziell.
Das ist nicht nur schlecht für den Wirtschaftsstandort Deutschland, da Hersteller ihre Standorte in andere Länder verlagern, sondern am Ende auch eine politische Verhinderung einer besseren Patientenversorgung.
Wir benötigen daher dringend weiterhin die Möglichkeit, Software der Risikoklasse I zu entwickeln und auch zuzulassen.
6. Fazit zur Risikoklasse I nach MDR
Nach all den Gerüchten und Diskussionen stand die Frage im Raum: „Gibt es noch Standalone-Software-Medizinprodukte der Risikoklasse I?“
Die Situation ist zwar nach viel Recherche und zahlreichen Gesprächen in unserem Umfeld weiterhin nicht final geklärt, allerdings lassen sich einige Erkenntnisse gewinnen:
-
- Aufsichtsbehörden, benannte Stellen und Hersteller interpretieren die MDR unterschiedlich, weshalb bis heute kein klarer Konsens in Bezug auf die Risikoklasse I bei Software herrscht.
- Das offizielle Update der MDCG-Guidance hat wenig in Bezug auf die bisherige Unklarheit zwischen Risikoklasse I und IIa geändert. Das ist eine sehr gute Nachricht. Denn das bedeutet unterm Strich: Es wurde zumindest nicht gewollt, dass Risikoklasse I z. B. für DiGA und Übungs-Apps explizit ausgeschlossen wird. Es ist davon auszugehen, dass Risikoklasse I auch in Zukunft von den meisten Aufsichtsbehörden in Bezug auf risikoarme Übungs-Apps akzeptiert wird. Ob das BfArM sich hierzu irgendwann eine andere Meinung bildet, bleibt abzuwarten.
Wir von QuickBird Medical sind der Meinung, dass es unbedingt Softwareprodukte der Risikoklasse I geben muss. Die Risiken, die von solchen Produkten ausgehen, sind verschwindend klein, haben aber in der Masse einen riesigen Einfluss auf eine bessere Patientenversorgung. Eine höhere Risikoklasse senkt die Attraktivität erheblich, solche Produkte zu entwickeln und zuzulassen. Die damit einhergehenden Aufwände sind für viele Unternehmen nicht zu stemmen oder schlicht zu riskant. Unser Gesundheitssystem – und vor allem Patienten – profitiert aber enorm von Produkten der Risikoklasse I.
Abonnieren Sie unseren Newsletter, um zur Risikoklassifizierung auf dem neuesten Stand zu bleiben.
Sprechen Sie uns auch gerne an, wenn Sie die Realisierung eines Software-Medizinprodukts planen. Wir entwickeln regulierte Apps, Web-Anwendungen und andere medizinische Software auf Auftragsbasis für andere Unternehmen.
The post Klasse I Software nach MDR – Geht das noch? (Dezember 2025) appeared first on QuickBird Medical.
]]>The post Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden appeared first on QuickBird Medical.
]]>Update – Juni 2025: Im Juni 2025 wurde das offizielle Guidance-Dokument „MDCG 2019-11: Guidance on Qualification and Classification of Software […]“ in Revision 1 angepasst. Alle Änderungen werden in diesem Artikel vollumfänglich berücksichtigt. Eine detaillierte Auflistung aller Änderungen finden Sie in dem unten stehenden Kapitel: Link zum Kapitel.
Update – Dezember 2025: Im Dezember 2025 hat die Europäische Kommission einen Entwurf zur Änderung der MDR vorgelegt, der unter anderem die Regel 11 zur Einstufung von Software-Medizinprodukten betrifft. Die potenziellen Folgen für Software der Risikoklasse I werden in Kapitel 9 dieses Beitrags eingeordnet.
- 1. Ist Ihre Software überhaupt ein Medizinprodukt?
- 2. MDR-Risikoklassifizierung: kurz und knapp
- 3. Die Zweckbestimmung entscheidet
- 4. In welche Risikoklasse fällt meine Software?
- 5.Wer trifft die finale Entscheidung?
- 6. Auswirkungen der Risikoklassifizierung
- 7. Risikoklasse I, IIa & IIb = DiGA?
- 8. Update der offiziellen MDCG-Guidance (Revision 1) – Juni 2025
- 9. Änderungsentwurf der MDR (Dezember 2025): Auswirkungen für die Klassifizierung von Software
- 10. Software unter MDR vs. IVDR
- 11. Fazit
1. Ist Ihre Software überhaupt ein Medizinprodukt?
Die Zweckbestimmung entscheidet, ob Ihre Software überhaupt als Medizinprodukt einzustufen ist. Erst wenn das der Fall ist, können Sie sich mit der Risikoklassifizierung beschäftigen. Der Artikel 2 (1) der MDR definiert klar, welche Produkte als Medizinprodukte einzustufen sind. So kann eine App zur Analyse der Herzleistung zum Beispiel entweder als Fitnesstracker vorgesehen sein, oder einen Kardiologen bei der Diagnostik unterstützen. Nur im zweiten Fall gilt die App als Medizinprodukt. Als Hersteller entscheiden Sie selbst, welchen Zweck Ihre Software hat. Sie wollen wissen, ob Ihre Software ein Medizinprodukt ist? Dann erhalten Sie in unserem Leitfaden einen genaueren Überblick: zum Leitfaden.
2. MDR-Risikoklassifizierung: kurz und knapp
Die Quintessenz der MDR Klassifizierungsregeln in aller Kürze: Die MDR unterscheidet 4 Risikoklassen: I, IIa, IIb & III 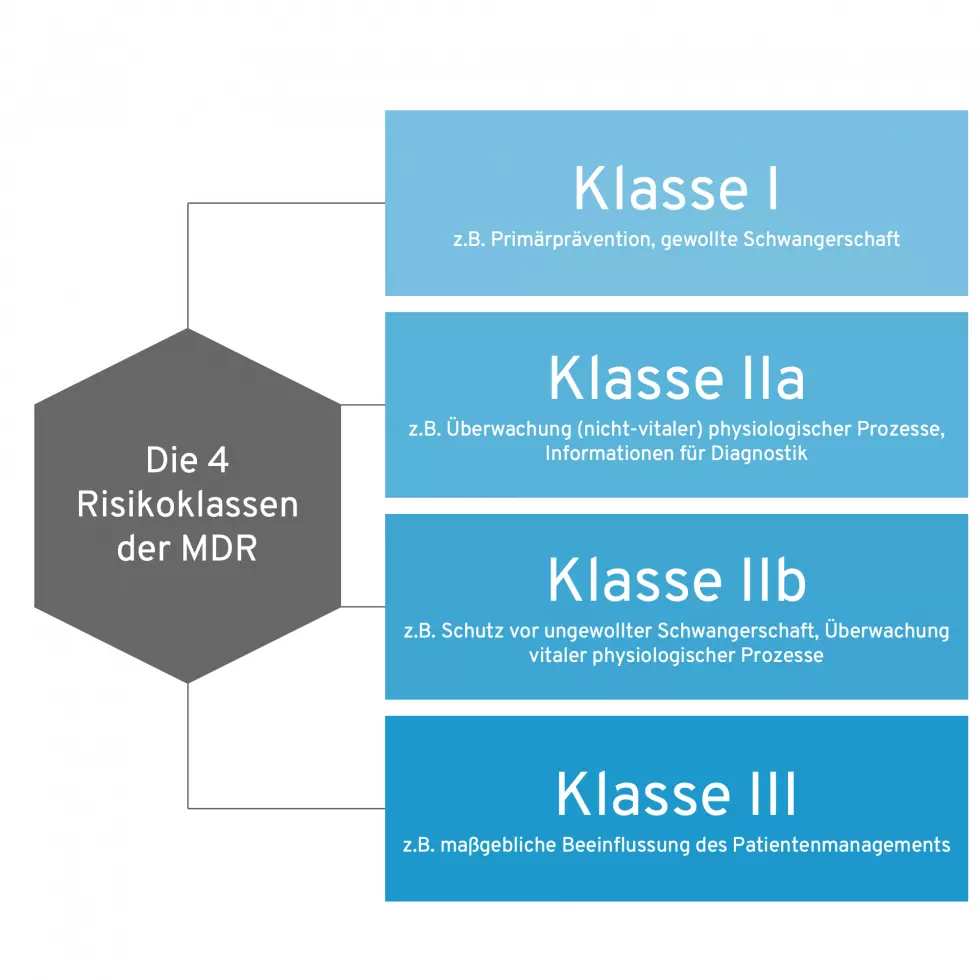
Die 4 MDR-Risikoklassen im Überblick
Als Daumenregel lässt sich sagen: Je größer der mögliche Schaden durch den Einsatz einer Medizinprodukt-Software ist, desto höher ist tendenziell die Klasse. Sobald Ihre Software Entscheidungen zu Therapie und Diagnostik eines Patienten beeinflusst, oder physiologische Prozesse (z.B. Schlafrhythmus) überwacht, fällt diese bereits mindestens in Klasse IIa (Regel 11). Wenn ein Patient dabei in großer Gefahr schwebt, steigt die Risikoklasse weiter an (z.B. Ihre App unterstützt nach einem Herzinfarkt). Eine Software, die vitale Parameter überwacht (Herz, Lunge, etc.) fällt ebenfalls in eine höhere Klasse. Sobald mehrere Regeln auf Ihre Software zutreffen, gilt die strengste Regel (die höchste Risikoklasse). Eine besonders hilfreiche Ergänzung zu diesem Leitfaden bietet das Guidance-Dokument der MDCG.
3. Die Zweckbestimmung entscheidet
Lassen Sie uns mit einem Beispiel beginnen, welches verdeutlicht, welchen Einfluss die medizinische Zweckbestimmung auf die Risikoklasse hat.
- Ihre App überwacht den Zyklus einer Frau, um die Wahrscheinlichkeit einer gewollten Schwangerschaft zu erhöhen. – Risikoklasse I
- Ihre App überwacht den Zyklus einer Frau, um das Risiko einer ungewollten Schwangerschaft zu verringern. – Risikoklasse IIb
Eine Steigerung um zwei Risikoklassen, auch, wenn es sich um ein und dasselbe Produkt handelt? Ja, tatsächlich macht die Zweckbestimmung einen immensen Unterschied. Demnach sollten Sie sich zunächst genau überlegen, wie die Zweckbestimmung Ihrer Software oder App lauten soll. Es kann gut sein, dass Sie Ihre Zweckbestimmung noch anpassen, nachdem Sie diesen Artikel zu Ende gelesen haben. Unser Artikel zur Zweckbestimmung beschreibt die genauen Inhalte und hilft Ihnen bei der Formulierung. Ob Ihre Software überhaupt ein Medizinprodukt ist, erfahren Sie in diesem Artikel.
3.1 Standalone-, Steuerungssoftware, und Zubehör
Jede Software mit einer eigenen medizinischen Zweckbestimmung wird gesondert klassifiziert. Insgesamt gibt es drei verschiedene Arten von Software, die wir zu Beginn unterscheiden möchten:
- Eigenständige Software / Standalone Software
- Software, die in Verbindung mit einem anderen Medizinprodukt verwendet wird
- Software als Teil eines Medizinprodukts
Wichtig an dieser Stelle ist für Sie, dass Sie Ihre Software nicht gesondert klassifizieren müssen, wenn
- sie integraler Bestandteil eines anderen Medizinprodukts ist (z.B. Embedded Software, wie Firmware eines Blutdruck-Messgeräts). Dies ist für mobile Apps fast nie zutreffend.
- sie ausschließlich zur Steuerung eines anderen Medizinprodukts dient (z.B. UI zur Bedienung eines Defibrillators). Dann fällt sie automatisch in dieselbe Klasse, wie das gesteuerte Produkt. Falls die Software aber noch eine weitere Zweckbestimmung hat, müssen Sie prüfen, ob sie in eine höhere Klasse fällt.
In allen anderen Fällen gilt es, die richtige Klasse für Ihre Software zu finden. Erfahren Sie hier mehr über die Arten von Software als Medizinprodukt.
4. In welche Risikoklasse fällt meine Software?
Wie bereits erwähnt, liefert die Zweckbestimmung Ihrer Software die Grundlage zur Frage, in welche der vier Risikoklassen sie fällt. Sobald die Zweckbestimmung klar ist, finden Sie mit den Regeln in Anhang VIII zur MDR die richtige Klasse für Ihre Software. Wir empfehlen, die Suche nach der Klasse bei Regel 11 zu beginnen, da diese speziell Software adressiert. Gleichzeitig lohnt sich ein Blick in das Guidance-Dokument der MDCG. Dessen Kerninhalte sind in diesem Artikel zusammengefasst.
4.1 Die Regel 11 der MDR
Für medizinische Software ist in den meisten Fällen die Regel 11 entscheidend. Hier wird zwischen Software unterschieden, die Informationen für Therapie- und Diagnostik-Entscheidungen liefert, und jener, die physiologische Prozesse überwacht. Eine Software kann natürlich auch mehrere Zwecke haben. Gemäß Regel 11 müssen Sie folgende Entscheidungen treffen:
4.1.1 Ist Ihre Software dazu bestimmt, Informationen zu liefern, die zu Entscheidungen für diagnostische oder therapeutische Entscheidungen herangezogen werden?

Wenn ja, beantworten Sie im Folgenden diese Fragen: Können diese Entscheidungen den Tod oder eine irreversible Verschlechterung des Gesundheitszustands herbeiführen? Wenn das zutrifft, fällt Ihre Software in die Risikoklasse III. 💡 Beispiel:
Ihre Software liefert durch Bildanalysen die Entscheidungsgrundlage für einen Notarzt, wie er einen Patienten mit akutem Schlaganfall behandeln soll.
Können diese Entscheidungen eine schwerwiegende Verschlechterung des Gesundheitszustands oder einen chirurgischen Eingriff herbeiführen? Wenn das zutrifft, fällt Ihre Software in die Risikoklasse IIb. 💡 Beispiel:
Ihre Software liefert die Entscheidungsgrundlage für einen Physiotherapeuten, ab welchem Punkt ein verletztes Knie wieder belastet werden kann. Eine Überbelastung könnte zu einer Operation führen.
Wenn Sie beide Fragen mit “Nein” beantworten können, fällt Ihre Software vorerst in die Risikoklasse IIa. 💡 Beispiel:
Ihre Software misst mittels Fragebogen den Schweregrad einer ADHS-Symptomatik bei Erwachsenen.
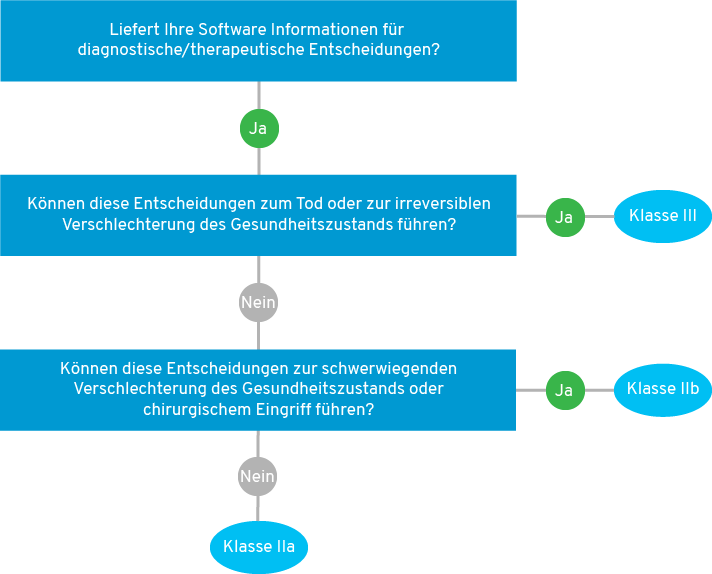
Grafische Darstellung des Entscheidungsbaums des ersten Teils der Regel 11.
4.1.2 Ist Ihre Software zur Überwachung von physiologischen Prozessen bestimmt?

Wenn ja, beantworten Sie im folgenden diese Fragen: Ist die Software zur Überwachung vitaler physiologischer Prozesse (Herz-, Lungen-, Hirnfunktion, etc.) bestimmt, wobei die Art der Änderung dieser Parameter zu einer unmittelbaren Gefahr für den Patienten führen kann? Wenn das zutrifft, fällt Ihre Software vorerst in die Risikoklasse IIb. 💡 Beispiel:
Ihre App überwacht die Herzfrequenz einer Person, während diese im Koma liegt. Bei gesundheitskritischen Messwerten wird das medizinische Personal alarmiert, um rechtzeitig eingreifen zu können.
Wenn Sie diese Frage mit “Nein” beantworten können, fällt Ihre Software vorerst in die Risikoklasse IIa. 💡 Beispiel:
Ihre App überwacht den Schlafrhythmus eines Patienten zu Hause, um den Erfolg einer Therapie von Schlafstörungen zu messen.

Grafische Darstellung des Entscheidungsbaums des zweiten Teils der Regel 11.
4.1.3 Hat Ihre Software einen anderen Zweck?
Die Regel 11 besagt im letzten Satz eindeutig: “Sämtliche andere Software wird der Klasse I zugeordnet.” Somit haben Sie auch für diesen Fall Ihre Antwort – oder doch nicht? Leider nein, denn dann stellt sich die Frage, ob eine andere Regel auf Ihre Software angewandt werden kann. Relevant sind dabei insbesondere die Regeln 15 und 22:
- Regel 15 richtet sich an aktive Produkte, die der Empfängnisverhütung oder dem Schutz vor sexuell übertragbaren Krankheiten dienen. Software in diesem Bereich fällt in Klasse IIb.
💡 Beispiel:
Ihre mobile App ist zur Empfängnisverhütung bestimmt. Dafür erinnert sie eine Nutzerin an die tägliche Einnahme der Anti-Baby-Pille. Wenn die Einnahme bis zu einer bestimmten Uhrzeit nicht bestätigt wurde, erzeugt die App ein Warnsignal.
- Regel 22 richtet sich an aktive therapeutische Produkte mit integrierter oder eingebauter diagnostischer Funktion, die das Patientenmanagement durch das Produkt erheblich bestimmt, wie etwa geschlossene Regelsysteme oder automatische externe Defibrillatoren. Software in diesem Bereich fällt in Klasse III.
💡 Beispiel:
Ihre Software ist zur weiteren Begleitung nach einer psychotherapeutischen Behandlung von Depression bestimmt. Durch die eingebaute Diagnostikfunktion wird die Behandlung durch die Software angepasst und die Notwendigkeit einer erneuten Psychotherapie bestimmt.
Wichtig: Wenn Ihre Software gemäß Regel 11 in die Risikoklasse IIb oder niedriger fällt, müssen Sie sich alle anderen Regeln ansehen – denn vielleicht ist noch eine strengere dabei, welche die Risikoklasse erhöht. Wenn mehrere Klassifizierungsregeln auf Ihre Software angewandt werden können, ist immer die strengste Regel zutreffend!
4.2 Was fällt überhaupt noch in Klasse I?
Hier existiert tatsächlich eine Grauzone bei der Interpretation der Regel 11 in der MDR. Wir geben detaillierte Hilfestellung in unserem Leitfaden zum Thema: Klasse I Software nach MDR – Geht das aktuell noch?.
4.3 Fallbeispiele zur Bestimmung der Risikoklasse
Diese Fallbeispiele sollen den Prozess der Entscheidungsfindung veranschaulichen. Es handelt sich hierbei um fiktive Beispiele.
4.3.1 Fallbeispiel 1: App zur Begleitung von Expositionstherapie
Ihre App soll die Expositionstherapie eines Patienten mit Höhenangst begleiten. Dazu wird gemeinsam mit seiner Therapeutin eine Liste mit Situationen erarbeitet, die dem Patienten Angst machen (z.B. 5 Minuten auf dem Balkon seiner Wohnung stehen.). Diese Situationen werden von “wenig angsteinflößend” bis “sehr angsteinflößend” geordnet. Nun soll er selbstständig zu Hause versuchen, eine dieser Situationen aufzusuchen und auszuhalten. Im Nachgang erfasst die App mittels Fragebogen, wie sich der Patient in der Situation gefühlt hat. Auf Basis dieser Daten entscheidet der Therapeut wöchentlich, wann der richtige Zeitpunkt für eine Steigerung des Schwierigkeitsgrades ist.  Wir beginnen bei Regel 11:
Wir beginnen bei Regel 11:
- Die App liefert Informationen, die therapeutische Entscheidungen beeinflussen. Wir könnten argumentieren, dass durch diese Entscheidungen weder der Tod, noch ein irreversibler oder ernsthafter Schaden entstehen kann. Demnach wäre das Produkt in Klasse IIa einzustufen.
Da keine andere Regel zutrifft, bleibt die App in Klasse IIa. Es ließe sich evtl. auch argumentieren, dass der Patient durch Therapieempfehlungen in eine Gefahrensituation gelangen könnte, die einen Tod hervorrufen kann (z.B. eine steile Klippe eines Berges). Somit würde das Produkt in Klasse III fallen. Wir entscheiden uns hier aber ausdrücklich, derartige Empfehlungen nicht in der App abzugeben. Dies zeigt jedoch gut den Argumentationsspielraum bei der Klassifizierung eines Medizinprodukts.
4.3.2 Fallbeispiel 2: App für die Therapie nach einem Herzinfarkt
Sie haben eine App entwickelt, die die Herzleistung eines Patienten nach einem Herzinfarkt zu Hause überwacht, nachdem die Behandlung abgeschlossen ist. Auf Basis der Messwerte informiert die App den Patienten, sobald die Einnahme eines lebensnotwendigen Medikaments notwendig ist und wann er wieder zur Nachsorgeuntersuchung gehen soll.  Wir beginnen bei Regel 11:
Wir beginnen bei Regel 11:
- Die App überwacht vitale physiologische Prozesse (Herzleistung). Demnach wäre die App ein Medizinprodukt der Klasse IIb.
Wir müssen uns auch die anderen Regeln anschauen, um herauszufinden, ob auch eine strengere Regel angewandt werden muss. Bei Regel 22 sehen wir:
- Ihre App passt die Therapie an und entscheidet selbst, welche Therapiemaßnahmen notwendig sind. Somit beeinflusst sie das Patientenmanagement.
Daher fiele Ihr Produkt in Risikoklasse III, weil immer die strengste Regel angewandt wird.
Achtung: Bei beiden Beispielen ließe sich sicherlich einen ganzen Nachmittag belebt diskutieren, ob diese Einschätzungen schlussendlich richtig sind. Wichtig für uns ist es hier, Ihnen einige greifbare Beispiele zum weiteren Verständnis zu geben. Eine glasklare Einstufung in eine Klasse ist nicht immer möglich, da das Gesetz unterschiedlich interpretiert werden kann. Es bleibt also Argumentationsspielraum. Genießen Sie diese Einschätzungen daher mit Vorsicht.
5. Wer trifft die finale Entscheidung?
Sie. Die Entscheidung, ob eine Software ein Medizinprodukt ist, trifft der Hersteller. Auch die Einstufung in eine Risikoklasse wird vom Hersteller vorgenommen. Dabei sollten Sie aber gewissenhaft vorgehen, denn die benannte Stelle kann die Zertifizierung verhindern, falls diese Ihrer Risikoklassifizierung nicht zustimmt. Hier erfahren Sie mehr über die Medizinprodukt-Zertifizierung: Zulassung & Zertifizierung von Software-Medizinprodukten (MDR) Bei einer zugewiesenen Risikoklasse I haben Sie zwar keine benannte Stelle, aber auch hier kann die zugewiesene Aufsichtsbehörde auf eine falsche Klassifizierung aufmerksam machen und Sie potenziell auffordern, Ihr Produkt nachträglich vom Markt zu nehmen. Wenn Sie unsicher sind, holen Sie am besten ein Gutachten oder eine Einschätzung einer dritten Partei ein. Die Möglichkeiten hierfür sind:
- Ein spezialisierter Anwalt, der Ihnen ein Gutachten ausstellt
- Ein Regulatory-Berater, der eine Einschätzung abgibt
- das BfArM (leider lange Antwortzeiten und daher kaum praktikabel)
Anmerkung: Weder eine Experteneinschätzung noch ein Gutachten schützen Sie rechtlich! Jedoch können solche Dokumente im Falle eines späteren Gerichtsverfahrens entscheidend sein.
6. Auswirkungen der Risikoklassifizierung
Die Risikoklassifizierung von Medizinprodukten hat entscheidenden Einfluss auf die regulatorischen Anforderungen und den Aufwand für die Zulassung und Marktüberwachung. Besonders der Übergang von Klasse I zu Klasse IIa ist hierbei kritisch, da mit der Höherklassifizierung erhebliche zusätzliche Anforderungen verbunden sind. Während Klasse-I-Produkte einer Eigenzertifizierung durch den Hersteller unterliegen, erfordert Klasse IIa die Einbindung einer Benannten Stelle, was die Dokumentationspflichten, Prüfverfahren und Kosten deutlich erhöht. Eine präzise und regelkonforme Risikoklassifizierung ist daher essenziell, um regulatorische Risiken zu minimieren und Verzögerungen im Marktzugang zu vermeiden. Was genau bei einem Risikoklasse IIa-Produkt im Vergleich zu Klasse I auf Sie zukommt, erfahren Sie in einem gesonderten Fachartikel von uns: MDR Risikoklasse I vs. IIa: Unterschiede für Software-Medizinprodukte
7. Risikoklasse I, IIa & IIb = DiGA?
Eine digitale Gesundheitsanwendung (DiGA) ist eine Software, die von ÄrztInnen und PsychotherapeutInnen verschrieben wird und deren Kosten von den gesetzlichen Krankenkassen übernommen werden. Damit eine Software oder App zur DiGA werden kann, muss sie in Risikoklasse I, IIa oder IIb fallen. Heißt das im Umkehrschluss, dass alle Software-Produkte, die diesen Risikoklassen angehören, auch automatisch DiGA sind? Nein! Um ins DiGA-Verzeichnis aufgenommen zu werden, müssen Apps einen dicken Anforderungskatalog erfüllen. DiGA sollen die Versorgung optimieren oder strukturelle Erleichterungen für PatientInnen schaffen. Software, deren Nutzer vorrangig ÄrztInnen sind, scheiden zum Beispiel bereits aus.  Zudem dürfen DiGA auch keine reinen Primärpräventionen darstellen. Da unter Risikoklasse I jedoch überwiegend solche Angebote fallen, sind auch deshalb viele Apps von der DiGA-Regelung ausgenommen. Alle Kriterien für eine DiGA finden Sie in diesem Artikel. Digitalen Medizinprodukten, die in Deutschland aufgrund der Risikoklasse III im Moment von der DiGA-Definition ausgenommen sind, wird nun in Frankreich eine Chance eröffnet. Alles über DiGA in Frankreich erfahren Sie in diesem Blogartikel.
Zudem dürfen DiGA auch keine reinen Primärpräventionen darstellen. Da unter Risikoklasse I jedoch überwiegend solche Angebote fallen, sind auch deshalb viele Apps von der DiGA-Regelung ausgenommen. Alle Kriterien für eine DiGA finden Sie in diesem Artikel. Digitalen Medizinprodukten, die in Deutschland aufgrund der Risikoklasse III im Moment von der DiGA-Definition ausgenommen sind, wird nun in Frankreich eine Chance eröffnet. Alles über DiGA in Frankreich erfahren Sie in diesem Blogartikel.
8. Update der offiziellen MDCG-Guidance (Revision 1) – Juni 2025
Im Juni 2025 wurde ein in der Digital-Health-Szene lange erwartetes Update des offiziellen Guidance-Dokuments der MDCG zur Risikoklassifizierung und Qualifizierung durchgeführt. Bisher war die MDCG-Guidance z.B. in Bezug auf patientenorientierte Übungssoftware (Großteil der im BfArM-Verzeichnis gelisteten DiGA) recht unkonkret. Es existierte eine relevante Grauzone bei der Interpretation der Regel 11 in Bezug auf die Einordnung in Risikoklasse I vs. IIa. Es wurde lange Zeit befürchtet, dass mit dem Update die Möglichkeit einer Risikoklasse I für Software-Medizinprodukte eliminiert werden könnte.
Entwarnung: Das ist glücklicherweise nicht passiert. Das neue Guidance-Dokument ist in Bezug auf Risikoklasse I vs. IIa z. B. bei patientenorientierter Software wie DiGA immer noch genauso unkonkret.
Persönliche Meinung von QuickBird Medical: Dass die Risikoklasse I für Patienten-Trainingsapps nicht explizit ausgeschlossen wurde, ist eine gute Nachricht. Eine Eingrenzung wäre ein schwerer Fehler gewesen. Sie hätte die europäische Innovation im Bereich digitaler Gesundheitsanwendungen massiv behindert. Risiken von Medizinprodukten müssen immer im Verhältnis zum Nutzen bewertet werden. Daher braucht es weiterhin Möglichkeiten, Apps unter Risikoklasse I zuzulassen. Nur so können auch kleine Unternehmen Innovationen auf den Markt bringen, von denen keine oder nur geringe Risiken ausgehen. Die Kosten und Aufwände eines Risikoklasse-IIa-Produkts sind für viele junge Unternehmen kaum finanzierbar. Nun zurück zur Guidance: Was hat sich also überhaupt geändert?
Konkrete Änderungen der Revision 1 im Juni 2025: Viel ist in Bezug auf Risikoklassifizierung tatsächlich nicht passiert. Hier sind die relevanten Änderungen zusammengefasst:
- Neues Beispiel für MDR Regel 11: Es wurde bezüglich der Klassifizierungsentscheidung mit Regel 11 der MDR ein neues Beispiel hinzugefügt: „[…] a device intended to prevent the risk of illnesses or pathologies by analysing physiological parameters (e.g. placement of the dorsal vertebrae, analysis of arterial stiffness, etc.) can be considered as a device providing information which is used to take decisions with diagnosis purpose (potential detection of pathologies) and in this case is in class IIa.“. Diese Klarstellung betrifft nur einen Bruchteil der am Markt befindlichen Software oder Apps, und ändert z.B. im Bereich von DiGA wenig bis nichts.
- Neuer Verweis auf MDCG-Guidance (Hardware-Software-Kombinationen): Es wird mehrmals auf die „MDCG 2023-4 Medical Device Software (MDSW) – Hardware combinations“ in Bezug auf Software-Hardware-Kombinationen verwiesen. Diese Guidance beinhaltet jedoch keine direkt relevanten Inhalte in Bezug auf die Risikoklassifizierung.
- Neuer Verweis auf MDCG-Guidance (Allgemeine Klassifizierung): Es wird auf die „MDCG 2021-24 for the general principles of medical device classification.“ verwiesen, welche sich mit allgemeinen Klassifizierungsregeln beschäftigt. Das ist unabhängig davon, ob Ihr Produkt eine Software darstellt oder nicht. Das ändert auch erstmal nichts Grundlegendes.
- Neue Informationen für Modularisierung: Das Kapitel über „Module“ wurde um weitere Informationen erweitert. Das könnte für Sie durchaus relevant sein, falls Sie bei der Unterteilung der Software in Medizinprodukt- und Nicht-Medizinprodukt-Komponenten Guidance benötigen.
- MDSW ersetzt Standalone Software: Der Begriff „Standalone Software“ wurde ersetzt durch den Begriff „Medical Device Software (MDSW)“. Dies wird damit erklärt, dass der Ort der Software (Embedded, auf einer Plattform, in der Cloud etc.) für die Klassifizierung nicht relevant sei.
Das war’s auch schon. Allgemein ergeben sich aus diesen Einschüben kaum Änderungen in Bezug auf die Medical Device Software (MDSW)-Klassifizierung.
9. Änderungsentwurf der MDR (Dezember 2025): Auswirkungen für die Klassifizierung von Software
Am 16. Dezember 2025 hat die Europäische Kommission einen Entwurf zur Änderung der Medical Device Regulation (MDR) vorgelegt. Ein wesentlicher Fokus dieses Vorschlags liegt auf einer Neufassung der Regel 11, die für die Risikoklassifizierung von Software-Medizinprodukten maßgeblich ist.
Auf den ersten Blick erscheint der neue Entwurf positiv und lässt vermuten, dass Software künftig grundsätzlich der Risikoklasse I zugeordnet wird, solange keine expliziten Kriterien für eine höhere Klassifizierung erfüllt sind. Diese scheinbar herstellerfreundliche Systematik relativiert sich jedoch bei näherer Betrachtung: Abhängig von der Auslegung könnte der Entwurf dazu führen, dass der überwiegende Teil medizinischer Software mindestens als Klasse IIa einzustufen wäre – auch bei vergleichsweise geringem Risiko.
Eine vertiefte Analyse des Entwurfs, der vorgeschlagenen Fassung der Regel 11 sowie der daraus resultierenden Interpretationsspielräume haben wir in einem separaten Beitrag zusammengefasst. Dort beleuchten wir insbesondere die konkreten Konsequenzen für Softwareprodukte: Zum Fachartikel
Wichtig für Hersteller ist: Der veröffentlichte Text stellt keine finale Rechtsfassung, sondern einen frühen Schritt im europäischen Gesetzgebungsverfahren dar. Erfahrungsgemäß werden solche Entwürfe im weiteren Verlauf weiter überarbeitet. Entsprechend ist derzeit nicht davon auszugehen, dass die vorgeschlagene Regelung unverändert Eingang in die endgültige Fassung der MDR finden wird.
10. Software unter MDR vs. IVDR
Falls Ihr Produkt z.B. auf Basis von Laborproben diagnostische Entscheidungen trifft, fallen Sie womöglich nicht unter die MDR, sondern unter die IVDR (In-vitro-Diagnostika-Verordnung).
Hier gelten komplett unterschiedliche Klassifizierungsregeln. Weitere Informationen zur Qualifizierung und Klassifizierung von IVDR-Software finden Sie in unserem Leitfaden: IVDR Software: Qualifizierung und Klassifizierung
11. Fazit
Um die richtige Risikoklasse für Ihre Anwendung zu finden, beginnen Sie am besten bei der Regel 11 der MDR. Fällt Ihre Software nach dieser Regel nicht in die Risikoklasse III, sollten Sie noch einmal alle anderen Regeln überprüfen, um herauszufinden, ob eine strengere Regel für Ihr Produkt anzuwenden ist. Zusätzlich lohnt sich ein Blick in das Guidance Dokument der MDCG, um anhand von weiteren Beispielen ein Gefühl für die Klassifizierung von medizinischer Software zu bekommen. Wichtig zu wissen ist, dass Sie als Hersteller die finale Entscheidung in der Frage nach der Risikoklasse Ihres Produkts treffen. Seien Sie sich dieser Verantwortung bewusst und holen Sie auch Meinungen von Experten ein.
Falls Sie eine Medical App, DiGA oder DiPA planen und Unterstützung bei der Entwicklung benötigen, setzen Sie sich gerne mit uns in Verbindung und wir besprechen das Ganze zusammen unverbindlich. Wir entwickeln Medical Apps für Unternehmen und Forschungseinrichtungen auf Auftragsbasis. Weitere Fragen? Rufen Sie uns gerne an (+49 (0) 89 54998380) oder schreiben Sie uns eine E-Mail ([email protected]). Abonnieren Sie unseren Newsletter für DiGA- und Medizinprodukt-Software-Hersteller, um auf dem Laufenden zu bleiben.
The post Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden appeared first on QuickBird Medical.
]]>The post Aktuelle DiGA Reports 2026: Offizielle Berichte im Überblick appeared first on QuickBird Medical.
]]>Für die strategische Planung von neuen DiGA-Projekten und Marktanalysen ist es wichtig, die Verschreibungszahlen gelisteter DiGA zu berücksichtigen. Bei der Suche nach aktuellen Statistiken stößt man allerdings auf unterschiedliche Berichte von verschiedenen Institutionen (GKV-Spitzenverband, Spitzenverband Digitale Gesundheitsversorgung, etc.).
Daher bieten wir in diesem Beitrag einen chronologischen Überblick mit Links zu den DiGA-Reports der relevantesten Herausgeber.
Dieser Artikel wird fortlaufend aktualisiert.
Inhalte dieses Artikels
- Alle DiGA Reports auf einen Blick
- Welche Daten nutzen die gelisteten DiGA-Reports?
- Welchen DiGA Report sollten Sie sich ansehen?
Alle DiGA Reports auf einen Blick
Jährlich werden die Abrechnungsdaten aller gesetzlichen Krankenkassen bis zum 30. September ausgewertet und in den hier aufgeführten Berichten dargestellt und zusammengefasst. Die Veröffentlichung der Berichte erfolgt meist im Frühjahr des folgenden Jahres.
DiGA Report 2024
- DiGA-Bericht des GKV-Spitzenverbandes – Spitzenverband Bund der Krankenkassen: Bericht mit den Versicherten-Daten aller Krankenkassen
- DiGA-Report 2024 – Marktentwicklung digitaler Gesundheitsanwendungen – Spitzenverband Digitale Gesundheitsversorgung (SVDGV): Bericht mit den Daten aller DiGA-Hersteller nach Selbstauskunft
DiGA Report 2023
- TK DiGA Report II – Der Bericht der Techniker Krankenkasse erschien im April 2024. Der Bericht bezieht sich, wie der nachfolgende Barmer Arztreport, auf Daten aus 2023
- Barmer Arztreport 2024
- DiGA-Bericht des GKV-Spitzenverbandes – Spitzenverband Bund der Krankenkassen
- Marktentwicklung digitaler Gesundheitsanwendungen (DiGA-Report) – Spitzenverband Digitale Gesundheitsversorgung (SVDGV): Bericht mit den Daten aller DiGA-Hersteller nach Selbstauskunft
DiGA Report 2022
- DiGA-Bericht des GKV-Spitzenverbandes – Spitzenverband Bund der Krankenkassen
- TK DiGA Report – Techniker Krankenkasse
DiGA Report 2021
- Digitalisierungsreport – DAK Gesundheit
Aktuelle Nachrichten zu DiGA und Medical Software finden Sie in unserem Newsfeed.
Welche Daten nutzen die gelisteten DiGA-Reports?
GKV-Spitzenverband:
Der GKV-Spitzenverband veröffentlicht seit 2022 jährlich einen DiGA-Report. Als Datengrundlage dienen die Abrechnungsdaten aller gesetzlichen Krankenkassen bis zum 30. September desselben Jahres (nach § 33a Abs. 6 SGB V).
Zudem definiert der GKV mit den Krankenkassen die Vergütungsbeträge für DiGA, die ab dem 13. Monat nach Aufnahme erstattet werden, und vereinbart darüber hinaus mit den DiGA-Herstellerverbänden (wie dem SVDGV) eine Rahmenvereinbarung, die u. a. Regelungen zu den Vergütungsverhandlungen, zu den Höchstbeträgen und zu den Schwellenwerten beinhalten.
SVDGV:
Mit dem diesjährig veröffentlichten DiGA-Report 2024 bietet der Spitzenverband Digitale Gesundheitsversorgung e.V. (SVDGV) erstmals Einblicke in die Entwicklungen des DiGA-Marktes auf Basis von Herstellerangaben. Die im Bericht erhobenen Daten wurden dementsprechend vom SVDGV einzeln bei jedem Hersteller für die jeweilige DiGA erfragt.
Da es sich um den ersten Report dieser Art handelt, umfasst er den gesamten Zeitraum nach der Listung der ersten DiGA vom Oktober 2020 bis September 2023.
2022 kritisierte der SVDGV den GKV-Spitzenverband scharf nach dessen Veröffentlichung des “DiGA Report 2022”. Laut SVDGV versäumte es der GKV damals erneut, den “noch jungen Versorgungsbereichs neutral darzustellen”. → zum Statement

Techniker Krankenkasse:
Deutschlands größte gesetzliche Krankenkasse mit über 11,3 Millionen Versicherten veröffentlichte bisher einmalig im Jahr 2022 einen expliziten DiGA-Report. Hierfür wurden Abrechnungsdaten von rund 16.000 TK-Versicherten ausgewertet, denen bis zum 31. Dezember 2021 eine DiGA verordnet oder genehmigt wurde.
BARMER:
In ihrem jährlich erscheinenden Arztreport setzt die Barmer erstmals 2024 ihr Schwerpunktthema auf DiGA und wertet hierzu die Daten ihrer rund 8,7 Millionen Versicherten aus. Zusätzlich wurden im November und Dezember 2023 insgesamt 1.000 Behandlerinnen und Behandler aus dem DocCheck Panel online über Einstellungen und Erfahrungen zu DiGA befragt.
DAK Gesundheit:
Die DAK veröffentlicht jährlich einen sogenannten Gesundheitsreport. Den letzten dezidierten „Digitalisierungsreport“, der letztlich auch das Thema DiGA umfasst, publizierte die in Hamburg ansässige Krankenkasse im Jahr 2021 in Kooperation mit der “Ärzte Zeitung”.
Als Datenbasis der zugrunde liegenden Studie dient hierbei eine Onlineumfrage vom 17. September bis zum 1. November 2021 sowie eine Befragung von ePatient Analytics mit 569 Ärztinnen und Ärzte als auch 16 Psychotherapeutinnen und -therapeuten aus ganz Deutschland.
Welchen DiGA Report sollten Sie sich ansehen?
Der DiGA-Report des GKV-Spitzenverbands bündelt zum jährlichen Stichtag am 30. September sämtliche Daten der gesetzlichen Krankenkassen und bietet dadurch mit Sicherheit die größte Datenmenge zur Inanspruchnahme von DiGA.
Allerdings sieht sich der GKV mit dem wiederholten Vorwurf des SVDGV konfrontiert, keine neutrale Berichterstattung zu liefern und DiGA zu delegitimieren, indem es die hohen Kosten von DiGA, das Zulassungsverfahren und die teils mangelnde Evidenz kritisiert. Das hatte diesjährig erstmals zur Folge, dass der SVDGV einen eigenen DiGA-Report veröffentlichte, ausschließlich basierend auf Herstellerangaben.
Die vereinzelten DiGA-(Zwischen-)Berichte der oben genannten Krankenkassen bieten ebenfalls interessante Erkenntnisse, allerdings begrenzt auf Daten der jeweils eigenen Versicherten.
Weitere Hilfestellungen und Marktanalyse-Tools für die Planung Ihrer DiGA:
- Blogbeitrag: Umsatz & Kosten: Lohnt sich eine DiGA? (inkl. Excel-Planner zur Prognose von Umsatz & Kosten Ihrer DiGA)
- DiGA-Verzeichnis: Analyse-Tool für Hersteller – Vergleich von DiGA anhand der Medizinprodukt-Risikoklasse, der Aufsichtsbehörde, der DiGA-Verordnungsdauer und dem Listen-Preis
- DiGA-Statistiken: Real-Time Dashboard – Informationen über die Risikoklassen-Verteilung, Preise bzw. Kosten, Dauer der Verordnungen u.v.m.
- Kostenloses Whitepaper: „13 wichtige Empfehlungen für angehende DiGA-Hersteller 2025“ – Erfahrungen aus unseren erfolgreich umgesetzten DiGA-Projekten zusammengefasst
The post Aktuelle DiGA Reports 2026: Offizielle Berichte im Überblick appeared first on QuickBird Medical.
]]>The post Alle Insights: DiGA-Entwicklung & Zulassung in 2026 appeared first on QuickBird Medical.
]]>Da wir DiGA auf Auftragsbasis für Start-ups, Pharma und andere Unternehmen entwickeln, sehen wir den kompletten Entwicklungszyklus einer DiGA: von der ersten Idee bis zur dauerhaften Zulassung und danach.
In diesem Beitrag möchten wir Ihnen einen Überblick über alle Themen geben, über die wir bereits geschrieben haben. Sie finden eine Übersicht aller Leitfäden und Fachartikel, eingeordnet nach Themengebiet.
Tipp: Wenn Sie neu in der Materie sind, macht es Sinn, dass Sie sich ganz einfach chronologisch von oben nach unten durch die Inhalte arbeiten.
Inhaltsverzeichnis
- 1. DiGA: Entwicklung & Zulassung
- 1.1 Ist Ihre App eine DiGA? Definition & Kriterien digitaler Gesundheitsanwendungen
- 1.2 12 wichtige Empfehlungen für angehende DiGA-Hersteller
- 1.3 Leitfaden zur DiGAV: Digitale-Gesundheitsanwendungen-Verordnung
- 1.4 DiGA: Leitfaden zum Nachweis positiver Versorgungseffekte
- 1.5 DiGA-Leitfaden: Historie aller Änderungen
- 1.6 Interoperabilität für digitale Gesundheitsanwendungen (DiGA)
- 1.7 Datensicherheits- & Datenschutz-Zertifikate für DiGA
- 1.8 DiGA & Software-Medizinprodukte ohne Qualitätsmanagement & Regulatorik zulassen?
- 1.9 BSI TR-03161 für DiGA: Zertifizierung der Datensicherheit
- 2. DiGA: Finanzen, Businessmodell, Marketing und Vertrieb
- 3. DiGA: Statistiken und Marktsituation
- 4. DiGA: Modelle in Europa
- 5. Medizinprodukt-Entwicklung und Zulassung
- 6. Alternative Erstattungsoptionen zu DiGA
- 6.1 Selektivverträge mit Krankenkassen: Die Alternative zu DiGA
- 6.2 Zertifizierung digitaler Kurse nach ZPP – Zentrale Prüfstelle Prävention
- 6.3 DiPA: Digitale Anwendungen für die Pflege
- 6.4 Erstattung von Software im GKV-Hilfsmittelverzeichnis (HMV)
- 6.5 Innovationsfonds und weitere Förderungen für Gesundheitssoftware
- 7. Weitere aktuelle Informationen zur Entwicklung von DiGA und Medizinprodukt-Software
1. DiGA: Entwicklung & Zulassung

Der Weg ins DiGA-Verzeichnis
Die folgenden Fachartikel & Leitfäden beschäftigen sich mit verschiedenen Aspekten der technischen und regulatorischen Entwicklung von DiGA:
1.1 Ist Ihre App eine DiGA? Definition & Kriterien digitaler Gesundheitsanwendungen
Zu Beginn Ihrer Planung stellt sich erstmal die Frage: Ist Ihre Anwendung überhaupt eine DiGA? Erfüllen Sie also alle Kriterien, um sich als DiGA zu qualifizieren?
1.2 12 wichtige Empfehlungen für angehende DiGA-Hersteller
Bevor Sie in die Entwicklung einer DiGA einsteigen, sollten Sie von Fehlern anderer DiGA-Hersteller lernen, um diese für Ihr eigenes Vorhaben zu vermeiden. Wir geben exklusive Einblicke und wichtige Empfehlungen aus der Praxis, die Sie bei Ihrer Planung beachten sollten:
1.3 Leitfaden zur DiGAV: Digitale-Gesundheitsanwendungen-Verordnung
Die DiGAV stellt die Grundlage für die gesetzlichen Anforderungen an digitale Gesundheitsanwendungen dar. Sie erhalten hier einen Überblick, was diese Anforderungen sind.
1.4 DiGA: Leitfaden zum Nachweis positiver Versorgungseffekte
In diesem Artikel geben wir Ihnen einen detaillierten Leitfaden darüber, wie der Nachweis positiver Versorgungseffekte zu erfolgen hat. Dies inkludiert Pilotstudie, Evaluationskonzept und die vollumfängliche Studie zur dauerhaften Aufnahme der DiGA.
1.5 DiGA-Leitfaden: Historie aller Änderungen
Der offizielle DiGA-Leitfaden ist wohl das wichtigste Dokument, das jeder DiGA-Hersteller gelesen haben sollte. Das BfArM aktualisiert diesen Leitfaden auch regelmäßig. Hierfür gibt es aber bisher leider keine detaillierte Änderungshistorie. Daher untersuchen wir alle Änderungen am DiGA-Leitfaden laufend stellen hier alle Änderungen mit Bildern und Kommentaren übersichtlich für Sie dar.
1.6 Interoperabilität für digitale Gesundheitsanwendungen (DiGA)
An eine DiGA werden gesetzlich eine Reihe von Interoperabilitätsanforderungen gestellt. In diesem Artikel erfahren Sie mehr über diese Anforderungen und erhalten praktische Einblicke in die Implementierung von Interoperabilitätsstandards für eine DiGA.
1.7 Datensicherheits- & Datenschutz-Zertifikate für DiGA
Digitale Gesundheitsanwendungen müssen strenge Datenschutz- und Datensicherheitsstandards erfüllen, um die Integrität und Vertraulichkeit patientenbezogener Daten zu gewährleisten. In unserem Artikel erhalten Sie einen umfassenden Überblick über die notwendigen Zertifikate und wie diese erlangt werden können.
1.8 DiGA & Software-Medizinprodukte ohne Qualitätsmanagement & Regulatorik zulassen?
DiGA und die Medical Device Regulation (MDR) konfrontieren Hersteller mit umfassenden Herausforderungen. Dies umfasst den Aufbau eines Qualitätsmanagementsystems (QMS), das Erstellen detaillierter technischer Dokumentation sowie das Management erhöhter regulatorischer Haftungsrisiken. In diesem Zusammenhang überlegen viele Unternehmen, ob sie die umfangreichen regulatorischen Anforderungen für Medizinprodukte vollständig an einen externen Anbieter delegieren können.
Dies ist tatsächlich vollständig möglich, indem die Rolle des Legalherstellers an ein externes Unternehmen ausgelagert wird.
1.9 BSI TR-03161 für DiGA: Zertifizierung der Datensicherheit
Seit dem 01.01.2025 müssen Hersteller digitaler Gesundheitsanwendungen (DiGA) die Erfüllung der Datensicherheitsanforderungen durch ein offizielles Zertifikat nachweisen. Als Basis der Zertifizierung wird die BSI TR-03161 herangezogen, welche 2020 erstmals veröffentlicht wurde.
Auf Basis unserer praktischen Erfahrungen aus mehreren durchgeführten TR-03161-Projekten schildern wir den aktuellen Stand der Zertifizierungsanforderungen.
2. DiGA: Finanzen, Businessmodell, Marketing und Vertrieb
Grafik zu den möglichen DiGA-Vetriebswegen
2.1 DiGA-Preisverhandlung mit dem GKV-Spitzenverband
In diesem Artikel wird beschrieben, wie Preisverhandlungen für DiGA mit dem GKV-Spitzenverband geführt werden. Er erklärt die Unterschiede zwischen der vorläufigen und der dauerhaften Aufnahme einer DiGA ins Verzeichnis und die damit verbundenen Zeitfenster für den Beginn der Verhandlungen. Der Artikel gibt einen Überblick über die notwendigen Unterlagen und die Dauer der Verhandlungen sowie die Rolle der Schiedsstelle bei nicht erzielten Einigungen.
2.2 Wie Sie DiGA-Daten für Vertrieb und R&D von Arzneimitteln nutzen können
Pharmazeutische Unternehmen ziehen großen Nutzen aus Real-World-Daten über die Zielgruppen ihrer Arzneimittel. Solche Daten sind besonders wertvoll für den Vertrieb bestehender Medikamente und die Entwicklung neuer Produkte.
Aber wie kann man mit einer DiGA Daten erheben, die dann z.B. für die Versorgungsforschung von Arzneimitteln verwendet werden können?
2.3 DiGA Umsatz & Kosten: Lohnt sich eine DiGA?
Beim Start einer DiGA ist es wichtig, das finanzielle Potenzial zu bewerten. Die zentrale Frage lautet: Ist die Entwicklung Ihrer DiGA-Idee finanziell sinnvoll? Dies fragen sich auch potenzielle Investoren. Sie sollten daher überzeugende Kalkulationen präsentieren, die Gewinn, Umsatzpotential, Marktpotenzial und Kosten umfassen. Dieser Artikel bietet Ihnen eine detaillierte Anleitung und Beispielrechnungen, um das Potenzial Ihrer DiGA für die gewählte Krankheitsindikation genau zu ermitteln.
2.4 Leitfaden: DiGA-Marketing und Vertrieb
Auch wenn alle regulatorischen Anforderungen erfüllt sind und Ihre DiGA im zentralen Verzeichnis gelistet wird, ist ein erfolgreicher Markteintritt damit noch nicht garantiert. Der bloße Eintrag bedeutet nicht automatisch, dass Ärzte Ihre DiGA verschreiben werden. Deshalb ist die Auswahl effektiver Marketing- und Vertriebsstrategien entscheidend, um das gesamte Marktpotenzial Ihrer Nische zu nutzen. Die Bandbreite der Möglichkeiten ist groß und das Marktumfeld komplex. Dieser Artikel bietet Ihnen einen Überblick und praktische Tipps.
3. DiGA: Statistiken und Marktsituation
3.1 Das DiGA-Verzeichnis für Hersteller
Digitale Gesundheitsanwendungen (DiGA) lassen sich anhand verschiedener Kriterien wie der Medizinprodukt-Risikoklasse, der zuständigen Aufsichtsbehörde, der Dauer der DiGA-Verordnung oder des Listenpreises vergleichen. Unser DiGA-Analyzer bietet im Gegensatz zum BfArM-Verzeichnis speziell für Hersteller angepasste Suchfilter, die eine gezielte Marktanalyse ermöglichen.
→ Link zur DiGA-Verzeichnis-Analyse

DiGA-Statistiken: Aktuelle gelistete DiGA
DiGA Statistiken: Das DiGA-Verzeichnis in Zahlen
Analysieren Sie das DiGA-Verzeichnis mithilfe unseres Echtzeit-Dashboards. Entdecken Sie aktuelle Informationen zur Verteilung der Risikoklassen, den Preisen und Kosten, der Verordnungsdauer und weiteren wichtigen Statistiken über die DiGA-Landschaft.
→ Link zu den DiGA-Statistiken
3.2 Aktuelle DiGA-Reports 2025: Offizielle Berichte im Überblick
Die aktuellsten Zahlen und Berichte für zugelassene digitale Gesundheitsanwendungen (DiGA) sind für die strategische Planung und Marktanalyse neuer DiGA-Projekte von großer Bedeutung.
Verschreibungszahlen und weitere Statistiken finden Sie in Berichten verschiedener Institutionen wie dem GKV-Spitzenverband oder dem Spitzenverband Digitale Gesundheitsversorgung.
Da die Informationen verstreut sein können, bieten wir Ihnen in diesem Beitrag einen chronologischen Überblick und verlinken direkt zu den DiGA-Reports der wichtigsten Herausgeber.
4. DiGA: Modelle in Europa
Deutschland hat 2019 Pionierarbeit geleistet, indem es ein standardisiertes Modell für die Zulassung digitaler Gesundheitsanwendungen (DiGA) einführte. In der Folge ziehen nun andere Länder in Europa nach und entwickeln ebenfalls DiGA-Modelle oder haben bereits solche implementiert.
In den folgenden Artikeln und Whitepapern werfen wir einen Blick darauf, für welche Länder …
… bereits ein DiGA-Modell existiert,
… ein DiGA-Modell geplant wird,
… oder keinerlei DiGA-Modell in Aussicht steht.
→ DiGA & DTX in Italien
→ DiGA & DTX in Österreich: Zulassung digitaler Gesundheitsanwendungen (2025)
→ DiGA & DT in der Schweiz
→ DiGA & DTX in Frankreich
→ DiGA & DTX in Belgien
→ DiGA & DTX in der EU: Der DiGA-Fast-Track – bald auch europaweit?
5. Medizinprodukt-Entwicklung und Zulassung
Vor der Zulassung als DiGA muss Ihr Produkt als Medizinprodukt nach Medical Device Regulation (MDR) zugelassen sein. Dies bringt zusätzlich zu den Anforderungen der DiGA-Verordnung viele Herausforderungen im Rahmen der MDR mit sich.
Wir haben eine Vielzahl von Fachartikeln geschrieben, welche sich speziell mit der Zulassung von Software-Medizinprodukten beschäftigen.
→ Link zur Übersicht über alle Artikel zur Medizinprodukt-Entwicklung und Zulassung
6. Alternative Erstattungsoptionen zu DiGA
In unserem Whitepaper „Alle Wege in die Kostenerstattung für medizinische Software und Gesundheits-Apps (in 2025)“ erhalten Sie einen Überblick über sämtliche Wege in die Kostenerstattung.

Neben der Zulassung als DiGA gibt es eine Reihe weiterer Wege, um DTx bzw. digitale Lösungen in die Erstattung durch Krankenkassen zu bringen. Nicht für jeden Hersteller ist der DiGA-Zulassungsprozess der beste Weg.
In den folgenden Artikeln gehen wir auf andere Erstattungsoptionen im deutschen Gesundheitssystem ein.
6.1 Selektivverträge mit Krankenkassen: Die Alternative zu DiGA
Selektivverträge sind eine spannende Alternative zum stark regulierten DiGA-Zulassungsprozess.
6.2 Zertifizierung digitaler Kurse nach ZPP – Zentrale Prüfstelle Prävention
Hersteller, die Produkte zur Krankheitsprävention entwickeln, sollten die Zertifizierungsmöglichkeiten durch die Zentrale Prüfstelle Prävention erwägen.
6.3 DiPA: Digitale Anwendungen für die Pflege
DiPA ist das Äquivalent zur DiGA im Pflegebereich. Erfahren Sie, wie Sie DiPA in die Erstattung bringen können.
6.4 Erstattung von Software im GKV-Hilfsmittelverzeichnis (HMV)
Softwareprodukte, die als medizinische Hilfsmittel gelten und beispielsweise körperliche Beeinträchtigungen ausgleichen, könnten in das Hilfsmittelverzeichnis aufgenommen werden. Dies eröffnet einen weiteren Weg zur Erstattung durch Krankenkassen.
6.5 Innovationsfonds und weitere Förderungen für Gesundheitssoftware
Für Hersteller digitaler Gesundheitslösungen existieren verschiedene öffentliche Förderprogramme – je nach Projektfokus, Reifegrad und Zielmarkt. Eine der zentralen und praxisrelevantesten Förderquellen im deutschen Gesundheitswesen ist dabei zweifellos der Innovationsfonds des G-BA, der gezielt neue Versorgungsformen unterstützt und gerade für Softwareprojekte mit Versorgungsbezug attraktive Möglichkeiten bietet.
7. Weitere aktuelle Informationen zur Entwicklung von DiGA und Medizinprodukt-Software
In die Leitfäden und Artikel ist viel Arbeit und Zeit geflossen. Wir hoffen, dass diese Ihnen als Hersteller bei Ihren Herausforderungen ein wenig helfen können.
Zuletzt können wir Ihnen noch unseren Newsletter empfehlen, wo wir monatlich einen detaillierten Bericht zu aktuellen Themen im Bereich der DiGA- und Medizinprodukt-Entwicklung versenden: Zum Newsletter
Aktuelle Entwicklungen und Nachrichten zum Thema DiGA & Medical Software finden Sie auch auf unserer News-Seite.
The post Alle Insights: DiGA-Entwicklung & Zulassung in 2026 appeared first on QuickBird Medical.
]]>The post Alle Insights: Entwicklung & Zulassung von Software-Medizinprodukten in 2026 appeared first on QuickBird Medical.
]]>In diesem Beitrag möchten wir Ihnen einen Überblick über die Themen geben, zu denen wir bisher geschrieben haben.
Tipp: Wenn Sie neu in der Materie sind, macht es Sinn, dass Sie sich ganz einfach chronologisch von oben nach unten durch die Inhalte arbeiten.
Inhaltsverzeichnis
- 1. Qualifizierung und Klassifizierung von Software-Medizinprodukten
- 1.1 Formulierung des Verwendungszwecks für (Software-)Medizinprodukte
- 1.2 Qualifizierung: Ist Ihre Software ein Medizinprodukt?
- 1.3 Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden
- 1.4 MDR-Risikoklasse I vs. IIa: Unterschiede für Software-Medizinprodukte
- 1.5 Klasse-I-Software gemäß MDR – Ist das noch möglich? (Januar 2025)
- 1.6 IVDR-Software: Qualifizierung und Klassifizierung
- 2. Leitlinien zu Aspekten der SaMD-Entwicklung und -Zulassung
- 2.1 Zulassung und Zertifizierung von Software-Medizinprodukten (MDR)
- 2.2 IEC 62304: Software-Lebenszyklusprozesse für Medizinprodukte
- 2.3 ISO 13485 – Leitlinien zu Anforderungen und Inhalten
- 2.4 Software als Medizinprodukt: 11 Zertifizierungstipps für Hersteller
- 2.5 Validierung von Medizinprodukt-Software gemäß MDR
- 2.6 MDR-Leitfaden: Klinische Bewertung von Software-Medizinprodukten
- 2.7 Künstliche Intelligenz (KI) in Medizinprodukten – MDR-Leitfaden (2026)
- 2.8 Zulassung von Software-Medizinprodukten ohne Qualitätsmanagement und Regulierung?
- 3. Erstattung und Vergütung für Software-Medizinprodukte
- 3.1 Digitale Gesundheitsanwendungen – unsere detaillierten Leitlinien
- 3.2 Selektive Verträge mit Krankenkassen: Die Alternative zur DiGA
- 3.3 Zertifizierung digitaler Kurse gemäß ZPP – Zentralstelle für Prävention
- 3.4 DiPA: digitale Anwendungen für die Pflege
- 3.5 Erstattung für Software, die in der GKV-Liste der Hilfsmittel (HMV) enthalten ist
- 3.6 Innovationsfonds und andere Förderungen für Gesundheitssoftware
- 3.7 Kostenerstattung für Software in Krankenhäusern
- 4. Weitere aktuelle Informationen zur Entwicklung von Medizinproduktesoftware und DiGA
1. Qualifizierung und Klassifizierung von Software-Medizinprodukten

Zweckbestimmung und Qualifikation von Medizinprodukten
1.1 Formulierung der Zweckbestimmung für (Software-)Medizinprodukte
Zu Beginn jeder Medizinprodukt-Entwicklung sollte die Zweckbestimmung formuliert werden, welche auch als Basis für die Zuordnung zur Risikoklasse nach MDR dient:
1.2 Qualifizierung: Ist Ihre Software ein Medizinprodukt?
Nachdem Sie die Zweckbestimmung formuliert haben, stellt sich die Frage „Ist Ihre Software überhaupt ein Medizinprodukt nach MDR?“. Wenn dies nicht der Fall ist, müssen Sie sich auch nicht weiter mit der MDR auseinandersetzen.
Lesen Sie diesen Artikel, um dies für Ihr Produkt herauszufinden.
1.3 Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden
Die Risikoklasse eines Medizinprodukts macht einen gewaltigen Unterschied für den notwendigen Aufwand und die Kosten, die Sie für die Zulassung investieren müssen. Sie sollten dies also vor Beginn der Produktentwicklung abklären.
1.4 MDR Risikoklasse I vs. IIa: Unterschiede für Software-Medizinprodukte
Gerade zwischen Risikoklasse I und Risikoklasse IIa existieren enorme Unterschiede für Kosten und Zeitplan im Rahmen der Produktentwicklung. Was sind die Unterschiede für Sie in der Praxis?
1.5 Klasse I Software nach MDR – Geht das noch? (Januar 2025)
Wir gehen darauf ein, welche Software-Medizinprodukte Stand 2025 noch in Klasse I fallen können.
1.6 IVDR-Software: Qualifizierung und Klassifizierung
Falls Ihre Software für den Informationsgewinn durch In-vitro-Untersuchungen bestimmt ist, fällt sie möglicherweise unter die IVDR (In-vitro Diagnostics Regulation). In unserem Leitfaden erfahren Sie, wie Sie Ihre Software unter die IVDR qualifizieren und klassifizieren können, um sicherzustellen, dass sie die relevanten regulatorischen Anforderungen erfüllt.
2. Leitfäden zu Aspekten der Entwicklung und Zulassung von SaMD

Timeline: Der Weg zur Medizinproduktzulassung
2.1 Zulassung & Zertifizierung von Software-Medizinprodukten (MDR)
Hier gehen wir einen holistischen Überblick über alle Aspekte der Zulassung von Software as a Medical Device (SaMD) ein.
2.2 IEC 62304: Software-Lebenszyklus-Prozesse von Medizinprodukten
Die IEC 62304 ist die wohl zentralste Norm zur Entwicklung von Software-Medizinprodukten. Doch wie genau lässt sich diese in die Praxis umsetzen und lässt sich dies mit agiler Entwicklung vereinen?
2.3 ISO 13485 – Leitfaden zu Anforderungen und Inhalten
Das Qualitätsmanagementsystem eines SaMD-Herstellers sollte mit der ISO 13485 im Rahmen der MDR im Einklang stehen. Dies stellt die wichtigste Norm für die Konformität von Qualitätsmanagementsystemen für Medizinprodukte dar.
2.4 Software als Medizinprodukt: 11 Zertifizierungs-Tipps für Hersteller
Seit über 10 Jahren entwickeln wir Software-Medizinprodukte auf Auftragsbasis für andere Unternehmen. Wir haben hier unsere wichtigsten Learnings und Tipps für zukünftige Hersteller aufgeschrieben.
2.5 Validierung von Medizinprodukt-Software nach MDR
Bevor ein Software-Medizinprodukt auf den Markt gehen kann, muss es formal validiert werden. Was das genau bedeutet, erfahren Sie hier.
2.6 MDR-Guide: Klinische Bewertung von Software-Medizinprodukten
Die klinische Bewertung ist das Kernstück der Validierung von Software-Medizinprodukt und stellt manch einen Hersteller vor große Herausforderungen. Wir gehen darauf ein, was Sie in diesem Rahmen tun müssen. Außerdem erklären wir, wann Sie eine klinische Studie/Prüfung durchführen müssen und wann eine Literaturrecherche ausreicht.
2.7 Künstliche Intelligenz (KI) in Medizinprodukten – MDR Leitfaden (2026)
Auch MDR-Medizinprodukte dürfen natürlich Künstliche Intelligenz (KI) enthalten. Doch die Zulassung derartiger Produkte bringt eine Reihe von zusätzlichen Herausforderungen mit sich. Hier gehen wir darauf ein, was Sie bei der Entwicklung von KI-Medizinprodukten beachten sollten.
2.8 Software-Medizinprodukte ohne Qualitätsmanagement & Regulatorik zulassen?
DiGA und die Medical Device Regulation (MDR) bringen für Hersteller eine Menge Herausforderungen mit sich – von der Einrichtung eines Qualitätsmanagementsystems (QMS) bis zur Erstellung der technischen Dokumentation. Kein Wunder, dass sich viele Unternehmen fragen, ob sie diese Komplexität auslagern können.
Die gute Nachricht: Ja, das geht! Indem die Rolle des Legalherstellers an einen externen Anbieter übertragen wird, lassen sich die regulatorischen Anforderungen komplett an eine dritte Partei auslagern.
3. Erstattung & Vergütung von Software-Medizinprodukten
Die Zulassung als Medizinprodukt nach MDR befähigt Sie, Ihr Produkt auf dem Markt zu vertreiben. Das heißt aber noch nicht, dass Sie damit auch Geld verdienen.
Gerade im deutschen Gesundheitssystem ist es oft ratsam, einen Weg in die Erstattung durch Krankenkassen für Ihr Produkt zu finden. Hierfür gibt es je nach Art des Software-Produkts eine Palette an Erstattungsoptionen.
Die folgenden Leitfäden geben Ihnen einen Überblick über wichtige Wege für Software-Produkte in die Erstattung.
3.1 Digitale Gesundheitsanwendungen – unsere detaillierten Leitfäden
Die Zulassung als DiGA stellt für Patienten-orientierte Applikationen bzw. DTx den wohl beliebtesten Weg in die Erstattung dar. Die Zulassung ist aber alles andere als einfach und bringt enorme regulatorische Hürden mit sich. Wir haben zu vielen Aspekten der Zulassung Fachartikel geschrieben, um Herstellern mit Zusatzinformationen zu unterstützen.
→ Link zu allen Insights zur DiGA-Entwicklung und Zulassung in 2025
3.2 Selektivverträge mit Krankenkassen: Die Alternative zu DiGA
Selektivverträge mit Krankenkassen eigenen sich für eine breite Palette von SaMD und sind ein populärer Weg in die Erstattung.
3.3 Zertifizierung digitaler Kurse nach ZPP – Zentrale Prüfstelle Prävention
Hersteller von Produkten, die sich auf die Prävention von Krankheiten konzentrieren, sollten sich mit der Zentralen Prüfstelle Prävention auseinandersetzen. Dies stellt einen möglichen Weg dar, die Kosten von derartigen Anwendungen (teilweise) zu vergüten.
3.4 DiPA: digitale Anwendungen für die Pflege
Das Pendant zur DiGA in der Pflege sind die DiPA. Wie Sie DiPA potenziell in die Erstattung bringen, erfahren Sie hier.
3.5 Erstattung von Software im GKV-Hilfsmittelverzeichnis (HMV)
Software-Produkte, die sich als Hilfsmittel qualifizieren und z.B. eine Fehlfunktion des Körpers ausgleichen, können potenziell ins Hilfsmittelverzeichnis aufgenommen werden. Dies stellt einen weiteren Weg in die Erstattung durch Krankenkassen dar.
3.6 Innovationsfonds und weitere Förderungen für Gesundheitssoftware
Für Hersteller digitaler Gesundheitslösungen existieren verschiedene öffentliche Förderprogramme – je nach Projektfokus, Reifegrad und Zielmarkt. Eine der zentralen und praxisrelevantesten Förderquellen im deutschen Gesundheitswesen ist dabei zweifellos der Innovationsfonds des G-BA, der gezielt neue Versorgungsformen unterstützt und gerade für Softwareprojekte mit Versorgungsbezug attraktive Möglichkeiten bietet.
Neben dem Innovationsfonds kommen weitere nationale und europäische Förderinstrumente in Betracht, die Entwicklungs-, Studien- oder Implementierungskosten reduzieren und den Markteintritt strategisch absichern können. Einen strukturierten Überblick über relevante Förderprogramme, typische Förderlogiken und strategische Einordnung bietet unser Whitepaper „Finanzierung und Förderung von Digital Health-Produkten“
3.7 NUB als Kostenerstattung für Software-Produkte im Krankenhaus
Könnte das NUB-Verfahren („Neue Untersuchungs- und Behandlungsmethoden“) ein Weg sein, um Software-Innovationen im Krankenhaus zu erstatten? Wir haben über 2400 NUB-Anträge analysiert – mit spannenden Ergebnissen zu den Chancen und Grenzen der Kostenerstattung digitaler Lösungen im Krankenhaus.
4. Weitere aktuelle Informationen zur Entwicklung von Medizinprodukt-Software und DiGA
In die Erstellung der Leitfäden und Artikel haben wir viel Arbeit und Zeit investiert. Wir hoffen, dass sie Ihnen als Hersteller bei Ihren Herausforderungen Unterstützung bieten können.
Als zusätzliches Angebot empfehlen wir Ihnen unseren Newsletter, in dem wir monatlich detaillierte Berichte zu aktuellen Themen im Bereich der und Medizinprodukt-Software- und DiGA-Entwicklung versenden.
Hier können Sie sich für den Newsletter anmelden: zum Newsletter
The post Alle Insights: Entwicklung & Zulassung von Software-Medizinprodukten in 2026 appeared first on QuickBird Medical.
]]>The post Medizinprodukte und die Schweiz: was Sie über MepV und MDR wissen müssen appeared first on QuickBird Medical.
]]>Ist ein Markteintritt in die Schweiz für MDR-Medizinprodukte ohne Weiteres möglich? Wie kommen Schweizer Hersteller mit Ihren Produkten in den EU-Markt? Und wofür benötigt man einen Bevollmächtigten?
In diesem Artikel klären wir diese und weitere Fragen zum Eintritt in den jeweils anderen Wirtschaftsraum: von der EU in die Schweiz und andersherum.
Wir fokussieren uns hier auf Software-Medizinprodukte, die Inhalte treffen aber ebenso auf andere Arten von Medizinprodukten zu.
Inhalt
- 1. Schweizer Medizinprodukteverordnung (MepV) vs MDR
- 2. Als EU-Hersteller auf den Schweizer Markt kommen
- 3. Als Schweizer Hersteller auf den EU-Markt kommen
- 4. Fazit
Hinweis: Da wir in diesem Artikel nicht auf jeden Sonderfall eingehen können, werden hier die Anforderungen beschrieben, die im Allgemeinen für Hersteller gelten. Wir raten aber dazu, sich mit der zuständigen Aufsichtsbehörde in Verbindung zu setzen, um Auskunft zu etwaigen Übergangsregelungen zu erhalten.
1. Schweizer Medizinprodukteverordnung (MepV) vs MDR
Zu Beginn müssen wir uns den rechtlichen Rahmen in der Schweiz und der EU ansehen, um zu verstehen, woran sich Hersteller in den jeweiligen Regionen halten müssen.
Die schweizerische Medizinprodukteverordnung (MepV) ist geltendes Recht für Medizinprodukte in der Schweiz. Zunächst klingt das so, als gäbe es komplett unterschiedliche Regelungen, als jene von der MDR definierten. Bei genauerer Betrachtung finden sich jedoch starke Parallelen, zahlreiche Verweise und sogar komplette Kopien der MDR in der MepV wieder.
Kein Grund zur Panik also – die wichtigsten Unterschiede und die Implikationen, die sich daraus für Sie als Hersteller ableiten, fassen wir in diesem Artikel zusammen.
1.1 Definition Medizinprodukt
Die Definition eines Medizinprodukts ist in MDR und MepV nahezu deckungsgleich – bis auf ein paar unterschiedliche Formulierungen. Somit gibt es im Bereich der Qualifikation keinen Mehraufwand für EU- und Schweizer-Hersteller, wenn sie ihren Markt auf die EU bzw. Schweiz erweitern möchten.
Wenn Sie noch nicht wissen, ob es sich bei Ihrem Produkt um ein Medizinprodukt handelt, lesen Sie unseren Leitfaden: Ist meine App ein Medizinprodukt?
1.2 Rahmenabkommen EU-Schweiz
Ein geltendes Rahmenabkommen zwischen der Schweiz und der EU wurde lange diskutiert, ist am Ende aber leider gescheitert. Eine genaue Erörterung der Gründe dafür liefert für Sie als Hersteller keinen wirklichen Mehrwert, daher werden wir dieses Thema nicht weiter behandeln. Wichtig ist an dieser Stelle nur:
- Die Schweiz gilt als Drittland im Sinne der MDR.
Das bedeutet noch konkreter:
- Sie brauchen einen Bevollmächtigten mit Sitz in der Schweiz oder der EU, um ein Medizinprodukt auf den jeweiligen Markt bringen zu können (sofern Sie keinen eigenen Sitz dort haben).
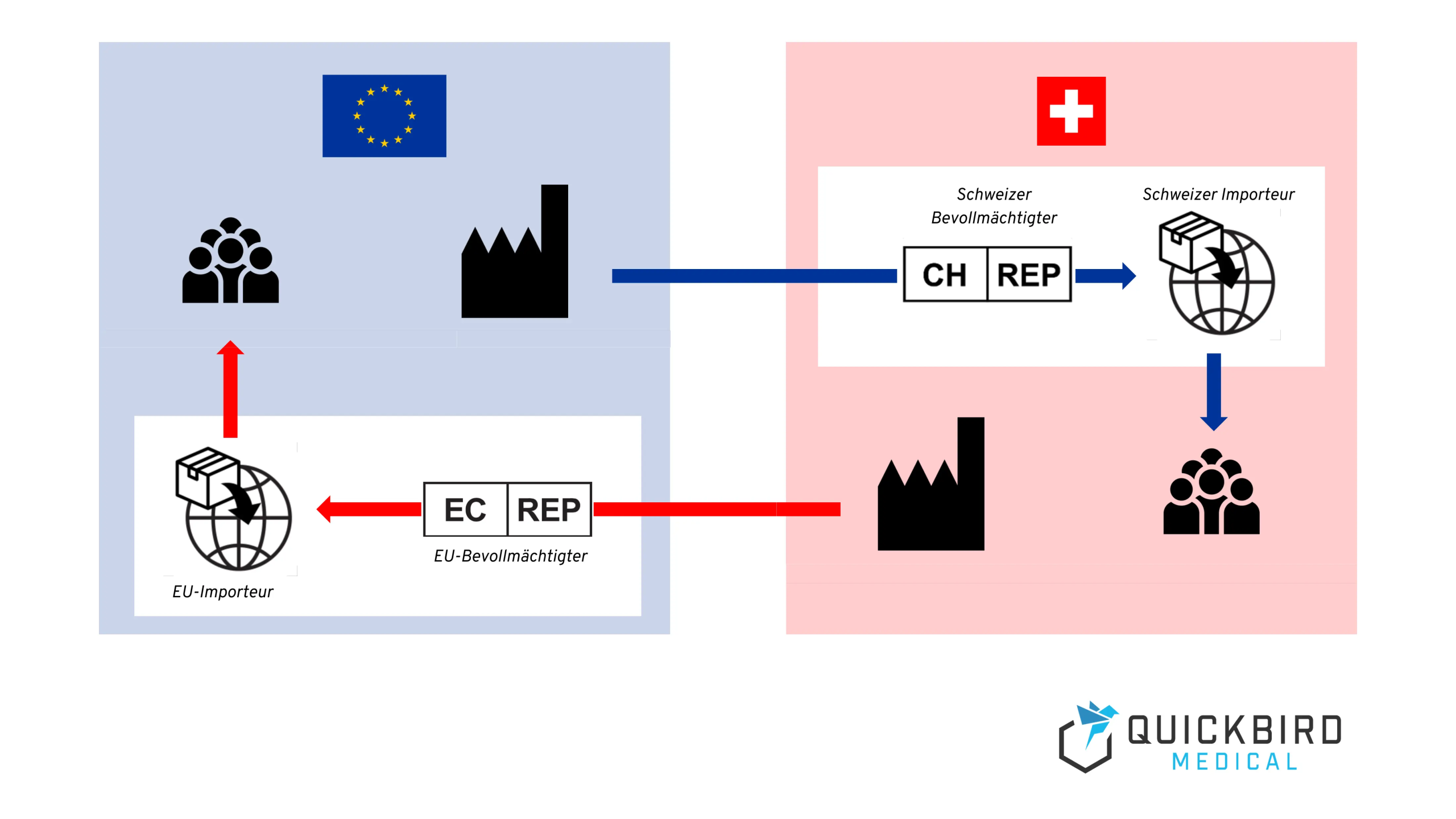
Das Rahmenabkommen der EU mit der Schweiz vereinfacht dargestellt
2. Als EU-Hersteller auf den Schweizer Markt kommen
Wenn Sie als Medizinprodukt-Hersteller nur in der EU ansässig sind und keinen Sitz in der Schweiz haben, besitzen Ihre Produkte bestimmt eine MDD- oder MDR-Zulassung. An dieser Stelle gibt es gute Nachrichten: Produkte, die eine CE-Kennzeichnung tragen, dürfen auch in der Schweiz auf den Markt gebracht werden und es gelten keine zusätzlichen Zertifizierungspflichten.
Die Swissmedic (schweizerische Zulassungs- und Aufsichtsbehörde für Arzneimittel und Medizinprodukte) schreibt auf ihrer Webseite explizit, dass Produkte mit einer CE-Kennzeichnung in der Schweiz in Verkehr gebracht werden können. Die Hauptfunktion der Swissmedic läge insbesondere in der Überwachung von Medizinprodukten, weniger in deren Zulassung.
Dennoch gibt es ein paar Dinge, die Sie tun müssen, um Ihr Produkt legal in der Schweiz vertreiben zu können:
- Ernennung eines Bevollmächtigten in der Schweiz
- Weisung des Bevollmächtigten und Klärung der Pflichten
- Anmeldung des Bevollmächtigten und Ihres Produkts in der Swissmedic Datenbank
- Veröffentlichung von Kurzberichten zur Sicherheit und Leistungsfähigkeit von Produkten (nur für Klasse III- und implantierbare Produkte)
- Bestimmung eines Importeurs in der Schweiz
2.1 Bevollmächtigter in der Schweiz
Wenn Ihr Unternehmen keinen eigenen Firmensitz in der Schweiz hat, müssen Sie einen Bevollmächtigten ernennen, welcher diverse Pflichten aus der MepV bzw. MDR für Sie übernimmt. Dieser Repräsentant muss eine natürliche oder juristische Person mit Sitz in der Schweiz sein.
Der Bevollmächtigte ist zuständig für die formellen und sicherheitsrelevanten Belange im Zusammenhang mit dem Inverkehrbringen des Produkts. Dazu zählt beispielsweise die Pflicht zur Meldung von Vorkommnissen und Trends, für die nach schweizerischem Recht immer der Bevollmächtigte verantwortlich ist (anders als in der MDR).
Wenn Sie die Pflichten des Bevollmächtigten in Artikel 51, MepV nachlesen, sehen Sie schnell, dass diese ansonsten (fast) deckungsgleich mit jenen der MDR sind. Dort wird nämlich schlichtweg auf Artikel 11 der MDR verwiesen.
Welche Aufgaben Sie an den Bevollmächtigten delegieren, bleibt zum Teil Ihnen überlassen, allerdings gibt es hier Einschränkungen. Nicht alle Herstellerpflichten können an den Bevollmächtigten abgegeben werden – dazu zählt beispielsweise die Durchführung einer klinischen Bewertung oder die Verantwortung für das Risikomanagement (alle Ausnahmen finden Sie in Absatz 4, Artikel 11 MDR).
Unabhängig davon hat der Bevollmächtigte die Pflicht, bei einer Anfrage der Behörde innerhalb von sieben Tagen dafür zu sorgen, dass diese die vollständige technische Dokumentation zu einem Produkt erhält. Der Schweizer Bevollmächtigte ist allerdings nicht dazu verpflichtet, eine Kopie dieser Dokumentation selbst zu haben – diese kann beim Hersteller bleiben.
Für die Rolle des Bevollmächtigten eignen sich beispielsweise Dienstleister, die diesen Service in der Schweiz anbieten.
2.2 Importeur in der Schweiz
Zusätzlich zu einem Bevollmächtigten, welcher gewisse Pflichten im Sinne der Medizinprodukt-Regulatorik übernimmt, benötigen EU-Hersteller auch einen Importeur in der Schweiz, um das Produkt dort in Verkehr zu bringen.
Die MepV definiert den Importeur als “jede in der Schweiz niedergelassene natürliche oder juristische Person, die ein Produkt aus dem Ausland auf dem Schweizer Markt in Verkehr bringt;”
Dieser hat ähnliche Pflichten, wie der Bevollmächtigte – im Detail nachzulesen sind diese in Artikel 13 und 16 der MDR bzw. Artikel 51 der MepV. Im Unterschied zum Bevollmächtigten benötigt er aber keine Person mit dem notwendigen regulatorischen Fachwissen im Medizinprodukte-Bereich. Ein Importeur übernimmt zum Beispiel aber auch bestimmte Pflichten in Bezug auf die Lagerung und den Transport von Medizinprodukten – da es sich bei (reiner) Software aber um kein physisches Produkt handelt, ist diese Anforderung bei Software-Medizinprodukten in der Regel nicht sinnvoll umsetzbar.
Auch der Bevollmächtigte selbst kann die Rolle des Importeurs übernehmen. In diesem Fall sind allerdings beide Rollen in der Swissmedic Datenbank anzumelden. Mehr dazu erfahren Sie im nächsten Kapitel.
2.3 Anmeldung in der Swissmedic Datenbank
Jeder Wirtschaftsakteur (Hersteller, Bevollmächtigter und Importeur) muss sich innerhalb von 3 Monaten, nachdem ein Medizinprodukt in der Schweiz in Verkehr gebracht wurde, in der Swissmedic Datenbank anmelden. Dabei werden Name, Anschrift und Kontaktdaten des Wirtschaftsakteurs, sowie der verantwortlichen Person gefordert. Nach der Registrierung erhält dieser eine CHRN – dabei handelt es sich um eine eindeutige Kennung. Sie ist das Äquivalent zur SRN in EUDAMED.
Der deutsche DiGA-Fast-Track – ein Modell für ganz Europa?
In diesem Whitepaper sehen wir uns an, in welchen EU-Ländern bereits ein DiGA-Modell existiert, ein DiGA-Modell geplant wird oder keinerlei DiGA-Modell in Aussicht steht. Außerdem klären wir die Frage: Kommt ein standardisiertes Modell in Europa für die EU-DiGA?
3. Als Schweizer Hersteller auf den EU-Markt kommen
Wenn Sie als Schweizer Hersteller ein Produkt auf den EU-Markt bringen wollen, müssen wir zunächst unterscheiden, ob Sie bereits ein Medizinprodukt haben, oder dessen Entwicklung erst bevorsteht. Beide Fälle beleuchten wir in den nachfolgenden Kapiteln.
WICHTIG: Als Schweizer Hersteller ohne Sitz in der EU benötigen Sie in jedem Fall einen Bevollmächtigten/Repräsentanten, sowie einen Importeur mit Sitz in der EU. Zudem müssen Sie Ihr Medizinprodukt in der EUDAMED Datenbank anmelden.
3.1 Sie haben noch kein Medizinprodukt
Bei der Entwicklung von Medizinprodukten für die EU gilt in erster Linie die MDR.
Da die MDR in der Schweiz anerkannt ist und die MepV an vielen Stellen darauf verweist, sind die Anforderung an die Produktentwicklung zum größten Teil identisch. Dazu zählt zum Beispiel der Aufbau eines Qualitätsmanagementsystems und die Erstellung einer technischen Dokumentation. Die genauen Anforderungen haben wir in anderen Blogartikeln bereits zusammengefasst:
- Leitfaden für die Entwicklung von Medical Apps: Darauf müssen Hersteller achten (MDR)
- Leitfaden: Ist Ihre App ein Medizinprodukt?
Die Frage, ob Sie zuerst den Schweizer- (unter MepV) oder den EU-Markt (unter MDR) betreten möchten, sollten Sie zu Beginn klären. Beides hat gewisse Vor- und Nachteile, welche Sie für Ihr Unternehmen abwägen müssen. Dazu müssen Sie unter anderem herausfinden, welcher Markt für Sie interessanter ist und inwiefern Sie Ihre technische Dokumentation offenlegen möchten (z.B. an einen EU-Bevollmächtigten).
Falls Sie bereits ein Medizinprodukt entwickelt und zugelassen haben, lesen Sie im nächsten Kapitel weiter.
3.2 Sie haben bereits ein Medizinprodukt nach MepV
Wenn Sie bereits ein Medizinprodukt konform mit der MepV entwickelt haben, haben Sie für das Produkt auch ein Konformitätsbewertungsverfahren durchgeführt. Bevor Sie das Produkt nun auf den EU-Markt bringen können, müssen Sie prüfen, ob das durchgeführte Verfahren auch für die EU zulässig ist. Sofern eine bezeichnete Stelle in die Konformitätsbewertung des Medizinprodukts involviert war, stellen Sie sicher, dass diese auch als benannte Stelle nach MDR anerkannt ist. In vielen Fällen ist dies nämlich nicht der Fall und Sie müssen ein weiteres Konformitätsbewertungsverfahren gemäß MDR durchführen.
Außerdem benötigen Sie einen Bevollmächtigten und einen Importeur mit Sitz in der EU und müssen Ihr Produkt in EUDAMED anmelden. Mehr dazu erfahren Sie in den folgenden Kapiteln.
3.2.1 Konformitätsbewertungsverfahren nach MDR
Um die CE-Kennzeichnung auf einem Medizinprodukt anbringen zu können, müssen Hersteller ein sogenanntes Konformitätsbewertungsverfahren durchführen. Die MDR bietet drei verschiedene Bewertungsverfahren, welche deckungsgleich mit den Verfahren der MepV sind. Bei Software-Produkten handelt es sich dabei meist um eine Bewertung auf Basis des Qualitätsmanagementsystems und der technischen Dokumentation. Zu diesem Thema finden Sie weitere Informationen in unserem Artikel: Zulassung & Zertifizierung von Software-Medizinprodukten (MDR)
3.2.2 Bevollmächtigter & Importeur in der EU
Genauso wie EU-Hersteller in der Schweiz, benötigen auch Schweizer Hersteller in der EU einen Bevollmächtigten (auch “Repräsentant”), um ihre Produkte auf dem EU-Markt vertreiben zu können.
Der Bevollmächtigte in der EU hat dieselben Pflichten, wie sein Äquivalent in der Schweiz (siehe weiter oben) – mit einer Ausnahme:
Der EU-Repräsentant muss über eine Kopie der technischen Dokumentation und der EU-Konformitätserklärung verfügen und diese auf Anfrage den zuständigen Behörden bereitstellen können.
Genauso wie in der Schweiz, benötigen Hersteller auch innerhalb der EU einen Importeur, welcher das Produkt in die EU importiert und auf dem Markt verfügbar macht.
Sie sind Medizinprodukt-Hersteller mit Sitz in der Schweiz? Dann sprechen Sie uns an – wir übernehmen gerne die Rolle des Bevollmächtigten und des Importeurs und stellen ein reibungsloses Inverkehrbringen Ihrer Produkte in der EU sicher.
3.3.3 Anmeldung in EUDAMED
Alle Wirtschaftsakteure (Hersteller, Bevollmächtigter und Importeur) müssen in EUDAMED registriert sein. Diese Anmeldung erfolgt über das entsprechende Online-Portal, nach welcher Sie eine SRN (eindeutige Kennung) erhalten.
Auch Medizinprodukte selbst müssen in der EUDAMED Datenbank registriert werden. Alles über die Zertifizierung und Zulassung von Medizinprodukten unter MDR erfahren Sie in unserem Artikel: Zulassung & Zertifizierung von Software-Medizinprodukten (MDR)
4. Fazit
Da aktuell kein Abkommen mehr zwischen der Schweiz und der EU besteht, wird die Schweiz im Sinne der MDR als Drittland behandelt. Das bedeutet, dass Medizinprodukt-Hersteller einen Bevollmächtigten/Repräsentanten in der EU brauchen, um ein Produkt dort in Verkehr zu bringen. Andersherum verhält es sich genauso, jedoch werden Medizinprodukte mit einer CE-Kennzeichnung in der Schweiz einseitig anerkannt, sodass für EU-Hersteller keine zusätzlichen Zertifizierungspflichten bestehen.
Schweizer Hersteller hingegen müssen häufig ein weiteres Konformitätsbewertungsverfahren (unter Einbezug einer benannten Stelle) durchführen, um Ihre Medizinprodukte in der EU anbieten zu können.
Mit Sitz in Deutschland übernimmt QuickBird Medical gerne die Rolle des Bevollmächtigten und des Importeurs innerhalb der EU. Sprechen Sie uns an, wenn Sie Unterstützung bei dem Inverkehrbringen oder der Entwicklung von MDR-Medizinprodukten benötigen. Wir helfen Ihnen gerne weiter.
The post Medizinprodukte und die Schweiz: was Sie über MepV und MDR wissen müssen appeared first on QuickBird Medical.
]]>The post Schweiz: Erstattung von Digitalen Gesundheitsanwendungen (DiGA & dGA) appeared first on QuickBird Medical.
]]>Dieser Prozess für die Zulassung von DiGA diente als Vorbild für Länder wie Frankreich und Belgien. Beide Länder entwickelten daran angelehnte Zulassungs- und Erstattungsprozesse. Auch in Österreich ist es geplant, DiGA in die reguläre Gesundheitsversorgung zu integrieren.
Bedingt durch ihre Lage ist auch ein Blick in die Schweiz interessant. Dort werden digitale Gesundheitsanwendungen „dGA“ genannt. Kommt das deutsche DiGA-Modell im nächsten Schritt auch in die Schweiz? Und welche Erstattungsmöglichkeiten für dGA gibt es dort jetzt schon?
Update Dezember 2025: Mit Beginn zum 1. Juli 2026 übernimmt die obligatorische Krankenpflegeversicherung in der Schweiz u.a. die Kosten für digitale Gesundheitsanwendungen zur kognitiven Verhaltenstherapie bei leichten bis mittelschweren depressiven Episoden. Mehr dazu finden Sie im unten stehenden Fachartikel.
Wir widmen uns in diesem Artikel
- den grundsätzlichen Eigenschaften des schweizerischen Gesundheitssystems
- der Frage, ob dGA in der Schweiz eine feste Definition haben
- den bisherigen Erstattungsmöglichkeiten und
- der Frage, ob ein DiGA-ähnlicher Zulassungsprozess in der Schweiz etabliert wird.
Inhaltsverzeichnis
- 1. Überblick: Aufbau des Gesundheitssystems und der Krankenversicherung der Schweiz
- 2. Anforderungen an DiGA
- 3. Definition von DiGA in der Schweiz
- 4. Aktuelle Erstattungsmöglichkeiten für DiGA
- 5. Zukunft: DiGA-Fast-Track bald in der Schweiz?
- 6. Fazit
1. Aufbau des Gesundheitssystems und Krankenversicherung
Zum besseren Verständnis, auf welches Umfeld DiGA in der Schweiz treffen, schauen wir uns zuerst an, wie das Gesundheitssystem dort funktioniert. Dies ist insofern von Bedeutung, als die historisch gewachsenen Gesundheitssysteme europäischer Länder selten miteinander vergleichbar sind: Unterschiede in Aufbau, Zuständigkeiten und Institutionen sowie Finanzierungssystemen erzeugen schlussendlich sehr unterschiedliche Anreizsysteme für alle beteiligten Gruppen.
Die Schweiz als Land in Europa, aber außerhalb der EU, ist hier keine Ausnahme.
1.1 Das Gesundheitssystem
Das schweizerische Gesundheitssystem ist dezentral organisiert. Während Gesetze und Vorschriften auf Bundesebene gelten, liegt die praktische Umsetzung bei den weitgehend autonom entscheidenden Kantonen: Ein Kanton in der Schweiz ist ein Gebiet mit eigener Regierung, eigenem Parlament und eigenen Gesetzen, das zusammen mit anderen Kantonen die Schweiz bildet.
Auch die Krankenversicherung ist kantonal sowie privatwirtschaftlich organisiert. Dies führt zu einer zersplitterten Gesundheitslandschaft, die jedoch für die Schweizer Bevölkerung im Allgemeinen eine sehr hochqualitative Versorgung bei gleichzeitig hoher finanzieller Eigenbeteiligung bietet.
Während auf Bundesebene vor allem die Gesetzgebung angesiedelt ist, können die Kantone die Umsetzung weitgehend selbst gestalten. Dadurch entsteht eine fragmentierte Gesundheitslandschaft.
1.2 Die Krankenversicherung
Seit 1996 gilt in der Schweiz eine Versicherungspflicht für die obligatorische Krankenpflegeversicherung (OKP). Die OKP deckt Gesundheitskosten, sobald die jährliche Mindestbeteiligung überschritten ist. Versicherte tragen weiterhin 10 % der Kosten bis zu einem Maximum von derzeit 700 Franken (Erwachsene) bzw. 350 Franken (Kinder) pro Jahr.
Die Beiträge variieren je nach Wohnort, Alter, gewählter Selbstbeteiligung (Franchise), Zusatzleistungen und Versicherungsmodell. Sie werden von den rund 40 Versicherern individuell festgelegt und jährlich vom Bundesamt für Gesundheit freigegeben. Arbeitgeber leisten keinen Beitrag.
Das Bundesamt für Gesundheit legt gültig für die gesamte Schweiz fest, welche Leistungen im Rahmen dieser Pflichtversicherung übernommen werden. Darüber hinaus bieten Krankenversicherungen eine Vielzahl an Zusatzversicherungen und Spezialtarifen an.
Die hohe Eigenbeteiligung der Versicherten, steigende Kosten und wachsende Prämien stellen das System vor große Herausforderungen.
2. Anforderungen an DiGA
In unserem Artikel „Medizinprodukte und die Schweiz: Was Sie über MepV und MDR wissen müssen“ finden Sie eine detaillierte Aufstellung dazu, welche Voraussetzungen Sie erfüllen müssen, um Ihr Software-Medizinprodukt in der Schweiz auf den Markt bringen zu können. Unberührt davon ist das Thema der Erstattung:
- Zulassung bedeutet, dass ein Medizinprodukt offiziell in der Schweiz verkauft und verwendet werden darf. Dafür muss das Produkt vereinfacht gesprochen nachweisen, dass es sicher ist und seinen medizinischen Zweck erfüllt. Die Zulassung wird durch gesetzliche Vorgaben geregelt. Mehr dazu finden Sie in unserem Leitfaden zur Medizinprodukt-Zulassung in der Schweiz.
- Erstattung bedeutet, dass etwa die Krankenversicherungen die Kosten für ein Produkt ganz oder teilweise übernehmen, sodass Patienten nicht (alles) selbst zahlen müssen.
In diesem Artikel geht es vor allem um Letzteres: alle Wege in die Erstattung für digitale Gesundheitsanwendungen in der Schweiz.
3. Definition von dGA: DiGA in der Schweiz
Vom Bundesamt für Gesundheit wurden DiGA im aktualisierten „Faktenblatt Vergütung von digitalen Gesundheitsanwendungen im Rahmen der OKP“ im November 2024 folgendermaßen definiert:
Unter digitalen Gesundheitsanwendungen (dGA) werden Produkte verstanden, welche zur Gesundheitsförderung, zur Unterstützung und Information von Patientinnen und Patienten, oder zu medizinischen Zwecken dienen und digitale Technologien nutzen.
Bei einer dGA kann es sich um eine Software, eine App, ein mobiles Gerät (z.B. Sensoren für die Erfassung von Körperparametern und Software zur digitalen Auswertung oder Übermittlung), oder auch um eine Kombination davon handeln. Auch kann der Einsatz von künstlicher Intelligenz (KI) zur Anwendung kommen. Ebenfalls kann eine dGA in Kombination mit einem nicht digitalen physischen Produkt (z.B. Personenwaage) zur Anwendung kommen. dGA können zur Selbstanwendung durch Patientinnen und Patienten oder gesunden Personen bestimmt sein oder zur Anwendung durch Fachpersonal.
Die Schweiz nutzt eine sehr breite Definition für digitale Gesundheitsanwendungen, die hier „dGA“ genannt werden.
Folgende Zwecke für eine dGA sind also möglich:
- Gesundheitsförderung (z. B. Fitness-, Ernährungs-, Bewegungs- oder Wellness-Apps)
- Unterstützung und Information (allgemeine, krankheitsbezogene Informationen und Verhaltensempfehlungen, ggf. mit Symptomtracking; wenn zur Information von Gesundheitspersonal gedacht, auch medizinischer Zweck denkbar)
- Medizinischer Zweck (Früherkennung, Verhütung, Diagnose, Überwachung, Prognose, Behandlung oder Linderung sowie Pflege von Krankheiten oder deren Folgen oder bei Mutterschaft)
Folgende Produktarten sind eingeschlossen:
- Software-Produkte
- Produkte, die Sensoren enthalten
- Produkte, die zusammen mit externer Hardware zur Anwendung kommen.
Nur für Produkte mit medizinischem Zweck ist dabei die Medizinprodukteverordnung (MepV) relevant.
Mehr zu verschiedenen Erstattungsmöglichkeiten von digitalen Gesundheitsanwendungen finden Sie im nächsten Kapitel.
4. Aktuelle Erstattungsmöglichkeiten für dGA
Nun interessieren Sie sich vermutlich dafür, ob DiGA im Rahmen des schweizerischen Gesundheitssystems vergütet werden können und ob es einen speziellen Zulassungsprozess gibt wie den deutschen DiGA-Fast-Track.
Digitale Gesundheitsanwendungen mit medizinischem Zweck werden in der Schweiz wie jedes andere (nicht-digitale) Medizinprodukt behandelt. Ein immer noch aktuelles Diskussionspapier aus dem Jahr 2022 skizziert folgende Möglichkeiten:
4.1 Vergütung innerhalb MiGel (Mittel- und Gegenständeliste)
DiGA können vom Bundesamt für Gesundheit (BAG) in die Leistungen der OKP (obligatorische Krankenpflegeversicherung) aufgenommen werden. Dabei handelt es sich um die Mindestversorgung, die Krankenversicherer ihren Versicherten anbieten müssen. Was zu dieser Mindestversorgung gehört, wird immerhin auf nationaler Ebene durch das BAG festgelegt und gilt somit in allen Kantonen.
Dafür müssten DiGA in die “Mittel- und Gegenstände-Liste” (MiGeL) aufgenommen werden, eine Art Hilfsmittelverzeichnis wie es auch in Deutschland existiert. Dort sind alle Hilfsmittel gelistet, die von der obligatorischen Krankenversicherung (OKP) übernommen werden. Beispiele wären Rollstühle oder Hörgeräte. Für jedes Produkt sind Produktgruppen, Preisobergrenzen, Mengen und Voraussetzungen für die Kostenübernahme aufgeführt.
Das MIGEL-Verzeichnis in der Schweiz ist für digitale Gesundheitsanwendungen nur bedingt geeignet, da die dort vorgesehenen Prozesse zur Abbildung von Innovationen zu langwierig sind. Sie dauern oftmals mehrere Jahre und stehen damit im starken Kontrast zu den kurzen Innovationszyklen digitaler Technologien.
Zusätzlich zur Einhaltung der Regeln nach MepV müssen Medizinprodukte für die Aufnahme in die MiGeL das sogenannte WZW-Prinzip erfüllen. Sie werden bei Antragstellung zur Vergütung nach OKP geprüft auf
- Wirksamkeit
- Zweckmäßigkeit
- Wirtschaftlichkeit

Das WZW-Prinzip nach KVG
Quelle: Kohler, S., & Rau, A. (2023, 26. April). Vergütung von digitalen Gesundheitsanwendungen (DiGA) in der Schweiz. VISCHER.
Aktuelle Entwicklungen: Hier kam es Ende 2025 zu den ersten positiven Entwicklungen. Mit Beginn zum 1. Juli 2026 übernimmt die obligatorische Krankenpflegeversicherung die Kosten für DiGA zur kognitiven Verhaltenstherapie bei leichten bis mittelschweren depressiven Episoden sowie bei wiederkehrenden depressiven Störungen, die als Ergänzung oder Überbrückung zur Psychotherapie eingesetzt werden. Die als erste in der MiGeL aufgeführte DiGA ist die in Deutschland bereits dauerhaft gelistete DiGA deprexis“.
4.2 Vertrauensprinzip und ärztliche Leistungen
Für DiGA, die im Zusammenhang mit ärztlichen Leistungen stehen, kommt das sogenannte Vertrauensprinzip zur Anwendung. Dieses besagt, dass bei ärztlichen Leistungen grundsätzlich davon ausgegangen wird, dass sie den gesetzlichen Anforderungen an Wirksamkeit, Zweckmäßigkeit und Wirtschaftlichkeit entsprechen.
Aber:
Derzeit bestehen noch Unklarheiten bezüglich der konkreten Anwendung dieses Prinzips auf digitale Anwendungen. Aktuell gibt es zudem keine spezifisch auf digitale Leistungen ausgerichteten Abrechnungsmodelle im ärztlichen Tarifwesen. Auch die Integration solcher Leistungen in bestehende ärztliche Tarife ist noch nicht abschließend geregelt.
4.3 Fallpauschalen
Fallpauschalen kommen bisher vor allem im stationären Bereich zur Anwendung, sind aber auch für den ambulanten Bereich vorgesehen. DiGA könnten Bestandteil eines umfassenden Behandlungsprogrammes sein, das pauschal vergütet wird. In solchen Fällen muss klar definiert sein, welche Leistungen Bestandteil der Pauschale sind und wie diese tariflich berücksichtigt werden.
DiGA könnten insbesondere dann eingebunden werden, wenn sie Bestandteil eines standardisierten Behandlungspfades sind, der auch ärztliche Verordnungen und fachliche Begleitung der Patienten umfasst.
4.4 Komplexpauschalen (Bundled Payments)
Komplexpauschalen fassen verschiedene Leistungen eines Behandlungspfades über mehrere Leistungserbringer hinweg zusammen und vergüten diese pauschal. Sie können insbesondere bei planbaren Eingriffen und der Versorgung chronisch Erkrankter eingesetzt werden. Es handelt sich um eine relativ neue Möglichkeit zur Leistungsabrechnung.
Dabei werden Behandlungspfade definiert, die unterschiedliche Leistungsbestandteile (z. B. ärztliche Leistungen, DiGA, begleitende Betreuung) umfassen.
Neue Vergütungsmodelle wie die Komplexpauschalen haben es jedoch schwer, weil die bestehenden Strukturen und fehlende Anreize der Tarifpartner diesen im Weg stehen. Hürden sind unter anderem die unzureichende Einbindung der Leistungserbringer, die doppelte Spitalfinanzierung, der Kontrahierungszwang – also die Pflicht der Krankenkassen, mit allen zugelassenen Leistungserbringern Verträge abzuschließen – sowie das Verbot von Mehrjahresverträgen und rechtliche Unsicherheiten.
4.5 Leistungspflicht in Evaluation
Gemäß Art. 33, Abs. 3 KVG kann der Bundesrat vorsehen, dass neue oder umstrittene Leistungen – auch digitale – im Rahmen einer befristeten Leistungspflicht unter bestimmten Bedingungen vergütet werden, solange ihre Wirksamkeit, Zweckmäßigkeit und Wirtschaftlichkeit noch nicht abschließend geklärt sind. Dieses Verfahren wird als „Leistungspflicht in Evaluation“ bezeichnet. Eine Checkliste des Bundes dient der Bewertung, ob eine DiGA in dieses Verfahren aufgenommen werden könnte.
4.6 Pilotprojekte im Rahmen des KVG
Nach Art. 59b E-KVG können Pilotprojekte initiiert werden, um neue Modelle zur Kostensteuerung, Qualitätssicherung oder Digitalisierung zu testen. Diese Projekte können gezielte Ausnahmen im KVG-Rahmen erhalten, sind jedoch zeitlich und räumlich begrenzt und unterliegen einer Evaluation.
Für deren Durchführung ist eine Einigung zwischen den beteiligten Tarifpartnern erforderlich, ebenso wie substanzielle Vorleistungen durch die Projektpartner. Die Ergebnisse solcher Pilotprojekte könnten als Grundlage für spätere gesetzliche Anpassungen dienen, insbesondere bei der Entwicklung neuer Vergütungs- oder Versorgungsmodelle.
4.7 Selbstzahler und Krankenzusatzversicherungen
Zuletzt sei noch die Möglichkeit erwähnt, eine DiGA schlicht als Medizinprodukt auf den Schweizer Markt zu bringen und individuell an Selbstzahler zu vermarkten oder mit zugelassenen privaten Krankenversicherern in Verhandlungen über eine Kostenübernahme im Rahmen privater Zusatzversicherungen zu treten. In einem Leitfaden des Digital Center Bülach (LinkedIn) wird jedoch darauf hingewiesen, dass die Kostenübernahme durch einen oder mehrere Krankenversicherer die Übernahme über die zuvor beschriebenen Wege ausschließt. Andersherum gilt dies genauso.
5. Zukunft: DiGA-Fast-Track bald in der Schweiz?
Nun stellt sich die Frage, ob sich die Schweiz von ihren Nachbarländern inspirieren lässt, einen gesonderten Zulassungs- und Erstattungsprozess für „dGA“ zu etablieren, Stichwort DiGA-Fast-Track.
Bei einem Blick in die Strategie eHealth Schweiz 2.0, ist jedoch von DiGA nichts zu lesen. “Digitale Anwendungen” sind im Rahmen der Strategie lediglich in Form des elektronischen Patientendossiers erwähnt. Dies scheint allerdings noch nicht den Vorstellungen des Gesetzgebers nach umgesetzt zu sein, da eine Weiterentwicklung im Sommer 2023 vom Bundesrat veranlasst wurde.
Auch im Anschlussprogramm DigiSanté: Förderung der digitalen Transformation im Gesundheitswesen werden keine Pläne zur Implementierung von speziellen Zulassungs- und Erstattungswegen für DiGA erwähnt. Zentrales Ziel des Programms ist, dass “der Rückstand der Schweiz in der Digitalisierung des Gesundheitswesens aufgeholt” wird.
Dennoch ist eine vorsichtige Öffnung der OKP für digitale Therapien erkennbar. Die geplante Aufnahme digitaler Anwendungen zur kognitiven Verhaltenstherapie in die Leistungspflicht ab dem 1. Juli 2026 zeigt, dass digitale Gesundheitsanwendungen zumindest indikationsspezifisch zunehmend als regelversorgungsfähig angesehen werden. Dies könnte mittelfristig auch die Diskussion um strukturiertere Zulassungs- und Erstattungswege für weitere digitale Anwendungen befördern.
6. Fazit: Erstattung von dGA
In der Schweiz sind digitale Gesundheitsanwendungen (DiGA) bisher nicht als eigenständige Kategorie in die obligatorische Krankenpflegeversicherung integriert, und es existieren keine speziellen Zulassungs- oder Erstattungswege wie der DiGA-Fast-Track in Deutschland. Erste Ansätze, digitale Gesundheitsanwendungen über Zusatzversicherungen oder spezielle Programme anzubieten, sind vorhanden, bleiben jedoch begrenzt. Mit den jüngsten Entwicklungen in Bezug auf Psychotherapie-Anwendungen, könnte sich aber langfristig die MiGeL als ein vielversprechender Erstattungsweg für DiGA-Hersteller in der Schweiz etablieren.
Über unseren Newsletter halten wir Sie zum Stand von DiGA in der Schweiz und anderen Ländern auf dem Laufenden.
Für einen Überblick über den Erstattungsprozess von digitalen Gesundheitsanwendungen in anderen Ländern der EU sehen Sie sich folgende Whitepaper und Fachartikel an:
- DiGA in Deutschland
- DiGA in Frankreich
- DiGA in Belgien
- DiGA in Österreich
- DiGA in Italien
- Überblick über Zulassung von DiGA in allen Ländern der EU
Außerdem gibt es eine breite Palette von öffentlichen Förderungen (siehe unser Praxisleitfaden zu Digital Health-Förderungen) und anderen Erstattungswegen (siehe unser Whitepaper zu Selektivverträgen) für Health-Software.
Über uns: Wir bei QuickBird Medical entwickeln digitale Gesundheitsanwendungen und medizinische Software auf Auftragsbasis für Kunden der Gesundheitsbranche. Hierfür sind wir nach ISO 13485 und ISO 27001 zertifiziert. Wenn Sie ein digitales Produkt im Gesundheitswesen planen, setzen Sie sich gern mit uns über das Kontaktformular in Verbindung.
The post Schweiz: Erstattung von Digitalen Gesundheitsanwendungen (DiGA & dGA) appeared first on QuickBird Medical.
]]>The post ISO 13485 – Leitfaden zu Anforderungen und Inhalten appeared first on QuickBird Medical.
]]>Die ISO 13485 ist eine Norm, welche Anforderungen an ein Qualitätsmanagementsystem für Medizinprodukt-Hersteller definiert. Im Kern dabei soll das Ziel verfolgt werden, sichere und funktionstaugliche Produkte zu entwickeln.
Doch wie genau soll das gelingen? Was ist ein prozessbezogener Ansatz? Was sind die Inhalte der ISO 13485 und wie setze ich sie in meinem Software-Unternehmen um?
In diesem Artikel beantworten wir die wichtigsten Fragen zu den Inhalten, Anforderungen und Kapiteln der ISO 13485 und zu Qualitätsmanagementsystemen im Allgemeinen.
Überblick
- 1. Qualitätsmanagementsystem – einfach erklärt
- 2. ISO 13485 – Leitfaden für Software-Hersteller
- 3. Frequently asked questions (FAQ)
- 4. Weitere relevante Normen für Software-Hersteller
- 5. Medizinprodukt-Zulassung ohne Qualitätsmanagement-System?
- 6. Fazit
1. Qualitätsmanagementsystem – einfach erklärt
Bevor Sie sich an die Umsetzung der ISO 13485 setzen, sehen wir uns vorab kurz an, was ein Qualitätsmanagementsystem eigentlich ist. Ein Qualitätsmanagementsystem beschreibt die Gesamtheit aller Prozesse in Ihrem Unternehmen, die dazu notwendig sind, um Produkte gemäß den Produktanforderungen umzusetzen.
Der Begriff Qualität beschreibt hier im Grunde, wie gut das Ergebnis (Produkt) mit den Produktanforderungen (z.B. Patientensicherheit, medizinischer Nutzen, Kundenzufriedenheit) übereinstimmt.
Lassen Sie uns mit einem stark vereinfachten Beispiel beginnen, welches den Sinn eines Qualitätsmanagementsystems (nach ISO 13485) greifbarer macht:
Sie möchten ein Skalpell herstellen, wobei Sie das Metall (Rohstoff) von einem Lieferanten beziehen. Sie suchen online nach Stahl und finden ein gutes Angebot auf ebay von einem anonymen Anbieter, bei dem Sie sofort zuschlagen. Mit Ihrem Schleifstein aus der Küche bringen sie den Klumpen in Form und fertig ist das Medizinprodukt.
Das Skalpell wäre nun scharf und einsatzbereit. Würden Sie sich damit am offenen Herzen operieren lassen? Hoffentlich nicht.
Unter Einhaltung der ISO 13485 wäre die Herstellung des Skalpells nicht so unkontrolliert möglich. Sie müssten nämlich in diesem abgespeckten Beispiel schonmal auf jeden Fall die folgenden Prozesse dokumentieren und natürlich auch einhalten:
- Ermittlung der Produktanforderungen – Zunächst müssen Sie identifizieren, welche konkreten Anforderungen an Ihr Skalpell gestellt werden. Das können z.B. Forderungen von (potenziellen) Kunden, oder auch gesetzliche Vorschriften sein. (Kapitel 7.2.1 der ISO 13485)
- Bewertung des Lieferanten, welcher das Metall liefert anhand relevanter Kriterien – ein Privatanbieter auf ebay würde dieser vermutlich nicht standhalten (Kapitel 7.4.1 der ISO 13485)
- Bewertung des eingekauften Rohstoffs – Sie müssen überprüfen, dass der Rohstoff, den Sie einkaufen, auch genau Ihren Anforderungen entspricht (Verifizierung) (Kapitel 7.4.3 der ISO 13485)
- Sicherstellung einer geeigneten Arbeitsumgebung – eine private Küche birgt zahlreiche unkontrollierbare Einflüsse, welche die Qualität des Produkts gefährden könnten (Kapitel 6.4 der ISO 13485)
- Sicherstellung geeigneter Arbeitsausrüstung – Ihr alter Schleifstein ist für Skalpelle vermutlich nicht geeignet (Kapitel 6.3 der ISO 13485)
- Sicherstellung der Kompetenz des Personals – Als Buchhalter bringen Sie ggf. nicht die notwendigen Qualifikationen mit, um Stahl zu bearbeiten (Kapitel 6.2 der ISO 13485)
- … und so weiter (Kapitel 4 bis 8 der ISO 13485)
Man sieht also, die ISO 13485 ist streng und kontrolliert so ziemlich alles, was einen Einfluss auf die Qualität des Produkts haben könnte. Unternehmen, die die ISO 13485 einhalten, sind also zwangsläufig darum bemüht, möglichst nichts dem Zufall zu überlassen, um die konstante Qualität Ihrer Produkte zu gewährleisten – eben einfach das, was auch schon der Name “Qualitätsmanagement” sagt.
Da die ISO 13485 aber eine sehr allgemeine Norm ist und für alle Medizinprodukthersteller gleichermaßen gilt, ist sie an vielen Stellen schwammig, kryptisch und vielleicht sogar unverständlich geschrieben – besonders für Software-Hersteller.
Was soll ein zum Beispiel ein Produktionsprozess für eine App sein? Muss ich mich in unserem Büro jetzt mit Kontamination beschäftigen? Und wie soll ich nachweisen, dass meine Software in sterilem Zustand ausgeliefert wird???
Fragen über Fragen – dieser Blogartikel soll Licht ins Dunkel bringen und die Umsetzung der ISO 13485 für Software-Hersteller erleichtern.
2. ISO 13485 – Leitfaden für Software-Hersteller
Wie eingangs beschrieben, ist die ISO 13485 eine Norm für alle Hersteller von Medizinprodukten. Ganz gleich, ob es um Skalpelle, Prothesen oder eben Softwareprodukte geht.
Deshalb möchten wir in diesem Leitfaden eine bessere Orientierung geben und pro Kapitel aufzeigen, was die zentralen Punkte für Software-Hersteller sind.
2.1 Prozessbezogener Ansatz
Die ISO 13485 verfolgt einen prozessbezogenen Ansatz. Wie der Name schon sagt, bedeutet das für Sie: Sie müssen Prozesse schreiben und anwenden.
Diese Prozesse erstrecken sich über den gesamten Lebenszyklus eines Medizinprodukts – Begonnen bei der Planung, bis hin zur Außerbetriebnahme und umfassen auch Bereiche, die nur indirekt mit dem Produkt zu tun haben.
Ein Prozess benötigt jeweils Eingaben und erzeugt Ausgaben, welche wiederum die Eingaben für andere Prozesse sind oder sein können.
Ein Beispiel für einen Prozess könnte Risikomanagement sein, welcher die in der Abbildung dargestellten Eingaben (z.B. Medizinische Zweckbestimmung) benötigt und bestimmte Ausgaben (z.B. Riskomanagement-Plan) erzeugt:

Der prozessbasierte Ansatz der ISO 13485
2.2 Erste Schritte – Starting from scratch
Wenn Sie zum ersten Mal eine ISO Norm aufschlagen, werden Sie vielleicht schlagartig darüber nachdenken, doch lieber eine klassische Lifestyle-App zu entwickeln, um den strengen Medizinprodukt-Vorschriften zu entgehen.
Doch auch wenn die ISO 13485 im ersten Moment überwältigend wirkt – so schlimm ist sie dann doch nicht. Sobald Sie verstanden haben, wie die Norm zu lesen ist, werden Sie es weitaus leichter haben, diese auch umzusetzen.
Hinweis: Wir empfehlen stark, für den Aufbau eines Qualitätsmanagementsystems einen erfahrenen Berater hinzuzuziehen.
Dieser kann nicht nur bei der Interpretation der Norm helfen, sondern erstellt mit Ihnen gemeinsam einen Projektplan und unterstützt Sie auch bei der Umsetzung der Norm speziell in Ihrem Unternehmen mit all seinen Besonderheiten.
2.3 Wie liest man die ISO 13485?
Die ISO 13485 ist an sich sehr angenehm zu handhaben – wenn man weiß, wie. Sie können jedes Unterkapitel aus den Hauptkapiteln 4, 5, 6, 7 und 8 im Grunde als eigene Anforderung betrachten, welche eine explizite Umsetzung erfordert. Das Inhaltsverzeichnis stellt daher schon die optimale Basis für ein entsprechendes Mapping dar.
Nun müssen Sie in der Theorie nur einmal von oben bis unten durchgehen und sich bei jedem Punkt überlegen, wie Sie diesen umsetzen.
Vor allem, wenn Sie keine Vorerfahrungen haben, empfehlen wir NICHT, das gesamte Qualitätsmanagementsystem von Grund auf neu zu schreiben. Viel zielführender ist es, etwas Geld in die Hand zu nehmen und sich entsprechende Templates zu kaufen. Diese finden Sie online, wobei wir an dieser Stelle keinen Anbieter empfehlen.
Natürlich sind eingekaufte Templates meist sehr generisch, sofern sie nicht eigens für Ihr Unternehmen durch eine Consulting-Firma erstellt wurden.
Daher werden Sie an einigen Stellen Anpassungen vornehmen müssen, damit die Prozesse auch auf Ihr Unternehmen anwendbar sind. Das Anpassen von Templates ist aber meist weniger fehleranfällig und führt zu besseren Ergebnissen, als der Starting-from-scratch Ansatz.
Bei der Anpassung von Templates kann Ihnen ebenfalls ein Berater helfen, damit Sie durch mehrere Review-Schleifen zu einem guten Ergebnis für Ihr Unternehmen kommen.
Stellen Sie unbedingt sicher, dass Sie auch wirklich alle Kriterien erfüllen, bevor Sie das Qualitätsmanagementsystem in die Praxis bringen und mit der Entwicklung Ihrer Medizinprodukt-Software beginnen.
Nun aber genug der einleitenden Worte – sehen wir uns die Inhalte der ISO 13485 genauer an.
2.4 Inhalte der ISO 13485
Die ISO 13485 umfasst insgesamt 8 Kapitel, wobei sich aus den ersten 3 keine konkreten Umsetzungsmaßnahmen ableiten lassen. Hier finden Sie den Anwendungsbereich der ISO 13485, Verweise auf andere relevante Normen sowie die Begriffsbestimmungen.
Deshalb fokussieren wir uns in diesem Leitfaden auf jene Kapitel, welche praktische Implikation für Sie als Hersteller beinhalten. Wie oben beschrieben, finden Sie alle umzusetzenden Anforderungen in den Kapiteln 4, 5, 6, 7 und 8.

Inhalte & Anforderungen der ISO 13485
Wichtig: Es ist möglich, einzelne Anforderungen aus den Kapiteln 6, 7 und 8 als “nicht anwendbar” zu deklarieren, wenn diese für Ihr Unternehmen nicht einzuhalten ist. Im Software-Umfeld umfasst dies zum Beispiel die besondere Anforderungen an sterile Medizinprodukte (7.5.5). Wenn Sie bestimmte Kapitel aber nicht anwenden, müssen Sie eine Begründung dafür anführen.
2.4.1 Kapitel 4 – Qualitätsmanagementsystem
In diesem Kapitel finden Sie “Allgemeinen Anforderungen” an Ihr Qualitätsmanagementsystem, sowie “Dokumentationsanforderungen”.
Kapitel 4 erfordert etwa die Identifikation und Umsetzung aller notwendigen Prozesse, die von Ihrem Unternehmen umzusetzen sind. Die meisten davon finden Sie in der ISO 13485 selbst, jedoch gibt es auch Prozesse aus anderen Normen und Regularien, die hier von Bedeutung sind (z.B. MDR, IEC 62304 oder ISO 14971).
Dazu ist es notwendig, die Rolle(n) Ihres Unternehmens im Markt zu verstehen, um das Anwendungsgebiet des Qualitätsmanagementsystems definieren zu können.
Zudem finden Sie in diesem Kapitel generelle Anforderungen an Ihre Prozesse und das Qualitätsmanagementsystem. Im Fokus hierbei steht die Aufgabe, die Wirksamkeit des Qualitätsmanagementsystems sicherzustellen. Dies geschieht etwa durch die Bereitstellung erforderlicher Ressourcen oder die Vergabe bestimmter Verantwortlichkeiten und Rollen.
Eine zentrale Eigenschaft des Qualitätsmanagementsystems ist die Pflicht, so ziemlich alles zu dokumentieren. Auch für die Lenkung (z.B. Erstellung, Freigabe, Sicherstellung der Verfügbarkeit, etc.) von Dokumenten und Aufzeichnungen finden Sie in Kapitel 4 entsprechende Anforderungen.
Eine weitere Forderung von Kapitel 4 ist die Erstellung eines Qualitätsmanagement-Handbuchs und einer Medizinproduktakte. Ersteres stellt sozusagen einen Überblick über das Qualitätsmanagementsystems dar. Dort wird der Anwendungsbereich klar definiert und die Prozesslandschaft dargestellt.
Das Qualitätsmanagement-Handbuch hat keinen klaren Produktbezug, sondern bezieht sich auf das Qualitätsmanagementsystem selbst.
Die Medizinproduktakte hingegen enthält (oder verweist auf) jene Dokumentation, welche die Konformität einzelner Medizinprodukttypen oder auch -gruppen darlegt. Inhalte sind beispielsweise die Zweckbestimmung sowie eine Liste von Produktanforderungen.
Sie sehen also, Kapitel 4 stellt in erster Linie grundlegende Regeln dar, auch welchen das gesamte Qualitätsmanagement aufbaut.
2.4.2 Kapitel 5 – Verantwortung der Leitung
In diesem Kapitel geht es vor allem um eines: Commitment.
Beispielsweise wird die Leitung dazu verpflichtet, die notwendigen Rahmenbedingungen für ein effektives Qualitätsmanagementsystem zu schaffen und die anwendbaren regulatorischen- und Kundenanforderungen zu identifizieren.
Dies soll etwa durch die Definition von Qualitätszielen, einer Qualitätspolitik und der Ernennung eines “Beauftragten der Leitung” erfolgen. Außerdem muss die Leitung für angemessene Kommunikationskanäle im Unternehmen sorgen (z.B. Slack, E-Mail, etc.).
Hinweis: “Leitung” bezieht sich meist auf das obere Management .
Zentral ist zudem die Pflicht zur Durchführung von Management Reviews (Managementbewertungen). Diese finden in einem definierten Intervall statt (z.B. jährlich) und haben das Ziel, die fortdauernde Eignung, Angemessenheit und Wirksamkeit des Qualitätsmanagementsystems sicherzustellen.
Diese Bewertung lässt sich nicht nach Gefühl vornehmen, deshalb muss diese Managementbewertung auch auf der Basis von Daten aus verschiedenen Quellen durchgeführt werden (z.B. Kundenfeedback). Aus der Managementbewertung sollen demnach Maßnahmen herauskommen, welche das Qualitätsmanagement und die Produkte selbst verbessern. Zudem soll der Bedarf an weiteren Ressourcen im Unternehmen ermittelt werden.
2.4.3 Kapitel 6 – Management von Ressourcen
MitarbeiterInnen, Fähigkeiten, Laptops, Git, … das alles sind nur einige Beispiele von Ressourcen, die Sie möglicherweise benötigen, um ein Software-Medizinprodukt zu entwickeln. Da es in Software-Unternehmen selten Lieferketten oder Fabrikhallen gibt, stehen vor allem die personellen und digitalen Ressourcen im Fokus, weshalb Kapitel 6 unter anderem ein starkes HR-Thema ist.
Hier wird besonders die Qualifikation von MitarbeiterInnen betont, welche durch Ihr Unternehmen sichergestellt werden muss. Das hat einerseits Auswirkungen auf die Selektion neuer MitarbeiterInnen, als auch auf das Training und die Weiterbildung bestehenden Personals.
Und auch wenn moderne Software-Unternehmen ohne festen Arbeitsplatz auskommen und deren MitarbeiterInnen lediglich einen Laptop als einzig notwendiges Arbeitsgerät bekommen, ist das Thema “Infrastruktur” nicht zu vernachlässigen.
Diese ist dann eben digital, aber dennoch zentral für die Entwicklung sicherer und funktionsfähiger Medizinprodukt-Software. Sie müssen Anforderungen an diese Infrastruktur ermitteln und dokumentieren.
Mit der Arbeitsumgebung und Kontamination hingegen müssen sich Software-Hersteller in der Regel nicht beschäftigen. Wichtig ist es aber, ein entsprechendes (kurzes) Statement für den “Ausschluss” dieser Anforderung zu dokumentieren. Dies wird einerseits von der Norm selbst, als auch von einigen Auditoren gefordert.
2.4.4 Kapitel 7 – Produktrealisierung
In Kapitel 7 wird es spannend, denn das ist der Teil, mit dem Sie sich am intensivsten beschäftigen werden, wenn Sie ein Qualitätsmanagementsystem nach ISO 13485 etablieren möchten.
Hier geht es nämlich um das eigentliche Produkt, das entwickelt werden soll – von der Planung bis zur Fertigstellung. Zunächst geht es darum, die Kundenanforderungen zu identifizieren und sicherzustellen, dass diese auch umgesetzt werden können.
Dazu zählen auch Anforderungen, die nicht explizit von Ihren Kunden gestellt werden (z.B. regulatorische Anforderungen).
In Kapitel 7 wird beschrieben, wie Sie die Produktrealisierung planen, die Produktanforderungen identifizieren und die Ergebnisse am Ende auch bewerten, verifizieren und validieren.
An dieser Stelle sei auf die IEC 62304 verwiesen, welche von Herstellern von Software-Medizinprodukten auf jeden Fall eingehalten werden sollte. Diese Norm beschreibt das Vorgehen bei der Software-Entwicklung präzise und stellt ergänzend zur ISO 13485 noch einmal zusätzliche Anforderungen an die Software-Entwicklung.
Die Produktentwicklung läuft nach ISO 13485 grob wie folgt ab:
- Entwicklungsplanung
- Entwicklungseingaben
- Entwicklungsergebnisse
- Entwicklungsbewertung
- Entwicklungsverifizierung
- Entwicklungsvalidierung
- Übertragung der Entwicklung an die Herstellung
- Lenkung von Entwicklungsänderungen
Die Reihenfolge der Kapitel suggeriert, dass diese Tätigkeiten streng aufeinander folgen. Das ist in der Realität aber nicht zwangsläufig der Fall. Einige Tätigkeiten können durchaus parallel ablaufen (z.B. Entwicklungsplanung und Ermittlung der Produktanforderungen (Entwicklungseingaben)).
Außerdem gibt es keine Forderung, dass all diese Tätigkeiten nur ein einziges mal durchlaufen werden. Im Hardware-Bereich macht das möglicherweise Sinn, aber in der agilen Software-Entwicklung ist es durchaus möglich, einige dieser Tätigkeiten in einem Zyklus laufen.
Bei der Realisierung von Software-Produkten ist zudem die IEC 62304 einzuhalten – Lesen Sie mehr dazu in diesem Artikel (IEC 62304: Software-Lebenszyklus-Prozesse von Medizinprodukten).
Hinweis: Was viele Hersteller verwirrt, ist die Unterscheidung zwischen “Entwicklungsverifizierung” und “Entwicklungsvalidierung”
Die Produktrealisierung umfasst nach der eigentlichen Entwicklung auch die Verifizierung und Validierung des Produkts. Da diese Unterscheidung oft unklar ist, hier eine kleine Faustregel, um den Unterschied besser zu verstehen. Der Unterschied lässt sich am besten anhand zweier Fragen festmachen:
- Verifizierung: Wurde das Produkt richtig umgesetzt?
- Validierung: Wurde das richtige Produkt umgesetzt?
Bei der Verifizierung gleicht man das Ergebnis mit den klaren Produktanforderungen ab – zum Beispiel “Die App zeigt dem Nutzer Therapieempfehlungen” (z.B. durch Software-System-Tests)
Bei der Validierung wird überprüft, ob das Ergebnis seinen Zweck auch erfüllen kann – zum Beispiel “Die Therapieempfehlungen lindern die Erkrankung” (z.B. durch eine klinische Studie).
Mehr zur Validierung erfahren Sie in diesem Artikel: Validierung von Medizinprodukt-Software nach MDR
Zudem finden Sie in Kapitel 7 Anforderungen an die Identifizierbarkeit und Rückverfolgbarkeit von Produkten, was Sie am besten über ein Versionierungssystem gewährleisten. Falls Überwachungs- und Messmittel verwendet werden (z.B. spezielle Testing Tools), sind auch diese gemäß ISO 13485 zu behandeln.
Beschaffung
Neben der eigentlichen Entwicklung des Produkts geht es in Kapitel 7 auch um die Regulierung eingekaufter Produkte und Dienstleistungen. Dabei geht es um die Bewertung von Lieferanten und die Verifizierung der eingekauften Leistungen.
Wenn Ihr Entwicklerteam beispielsweise mit Jira arbeitet, müssen Sie einerseits Atlassian (dessen Hersteller) als Lieferanten bewerten und anschließend untersuchen, ob Jira die von Ihnen gestellten Anforderungen an ein Ticket-Managementsystem erfüllt. Erst nach erfolgreicher Bewertung und Validierung dürfen Sie Jira in Ihrem Unternehmen einsetzen. Mehr zur Validierung von eingesetzter Software erfahren Sie in unserem Artikel: Validierung von Software nach MDR
2.4.5 Kapitel 8 – Messung, Analyse und Verbesserung
Sobald Ihr neues Qualitätsmanagementsystem im Einsatz ist, werden Sie bald sehen, dass die Prozesse an einigen Stellen noch nicht reibungslos funktionieren und vielleicht sogar zu regulatorischen Abweichungen führen.
Der Umgang mit Optimierungen und Korrekturen wird in Kapitel 8 behandelt – denn natürlich brauchen Sie auch hierfür eigene Prozesse. Doch nicht nur Daten über das Qualitätsmanagementsystem selbst, sondern natürlich insbesondere zu Ihren Medizinprodukten müssen entsprechend gesammelt und verarbeitet werden.
In diesem Kapitel finden Sie daher Kriterien für die Datenanalyse, Überwachung, Umgang mit nicht-konformen Produkten, sowie stetige Verbesserungen Ihres Qualitätsmanagementsystems.
Neben den regelmäßigen Überwachungsaudits sind Sie außerdem dazu verpflichtet, interne Audits durchzuführen, um die fortlaufende Konformität Ihres Qualitätsmanagementsystems sicherzustellen.
2.5 Update 2021 mit Amendment: ISO 13485:2016/A11:2021
2021 wurde die ISO 13485 durch die Änderung A11:2021 aktualisiert. Durch ISO 13485/A11:2021 wurde der Hauptteil der ISO 13485 nicht geändert. Neu hinzugekommen ist vor allem ein offizielles Mapping zur MDR. Dieses findet sich im Annex ZA.
Dieses Mapping ist bei der Analyse und Identifikation offener Anforderungen sehr hilfreich und sollte von Herstellern daher berücksichtigt werden.
Das heißt zusammengefasst, dass die Änderung A11:2021 keine neuen oder geänderten Anforderungen an das Qualitätsmanagementsystem stellt, sondern vor allem bei der Erlangung der MDR-Konformität unterstützt.
Da die neue Version harmoniert ist, sollten sie ab sofort die EN ISO 13485:2016/A11:2021 oder die DIN EN ISO 13485:2021 als Basis für Ihre MDR-Produkte verwenden.
2.6 QMS-Software, Atlassian und Co. – Das richtige Tooling
Sie sehen schon, ein Qualitätsmanagementsystem besteht aus zahlreichen Dokumenten und Aufzeichnungen, die in wechselseitiger Beziehung zueinander stehen. Ein sehr vernetztes System also, welches durch entsprechendes Tooling übersichtlicher gestaltet werden kann.
Qualitätsmanagementsystem klingt für viele Menschen immer noch nach sehr viel Papier. Natürlich kann man mit ausgedruckten Dokumenten arbeiten, und diese zum Beispiel mittels einer Unterschrift freigeben.
In der Regel ist dies aber nicht das Mittel der Wahl – gerade Software-Unternehmen, deren Mitarbeitende nicht immer an einem zentralen Ort arbeiten, stoßen mit diesem Ansatz auf Probleme.
Natürlich gibt es mittlerweile auch zahlreiche digitale Möglichkeiten, ein Qualitätsmanagementsystem umzusetzen. Ein paar Möglichkeiten zur Umsetzung sind in der untenstehenden Tabelle aufgeführt.
| Methode | Vorteile | Nachteile | Kommentar |
| Ausgedruckte Dokumente (z.B. Microsoft Word) |
|
|
Diese Methode ist besonders in älteren Unternehmen verbreitet. Vor allem in Software Unternehmen ist sie aber in den allermeisten Fällen nicht das Mittel zur Wahl. |
| Spezial-Software für QMS (z.B. Greenlight Guru) |
|
|
Spezial-Software kann in einigen Fällen sinnvoll sein – vor allem dann, wenn die Kosten erstmal keine große Rolle spielen. |
| Cloud-basierte Dokumentenmanagement-Software (z.B. Confluence/Jira) |
|
|
Diese Methode ist sehr flexibel und im Regelfall um einiges günstiger, als Spezial-Software für QMS. Daher ist sie besonders für Startups interessant. |
| Texteditor mit digitaler Versionsverwaltungs-Software (z.B. Word + Git) |
|
|
Auch diese Methode ist günstig und gut individualisierbar. Jedoch erfordert sie ein größeres technisches Verständnis als die anderen Methoden. |
3. Frequently asked questions (FAQ)
3.1 Wer braucht die ISO 13485?
Die ISO 13485 wird von Medizinproduktherstellern verwendet und beinhaltet Anforderungen an ein Qualitätsmanagementsystem. Ein solches wird beispielsweise von der MDR gefordert, um Medizinprodukte entwickeln zu können.
3.2 Was bestätigt ein ISO 13485 Zertifikat?
Ein ISO 13485 Zertifikat bestätigt, dass ein Unternehmen über ein Qualitätsmanagementsystem für Medizinprodukte verfügt. Ein solches ist in der gesetzlich vorgeschrieben, wenn Sie Medizinprodukte in der EU (und in vielen anderen Ländern) herstellen und vertreiben möchten. Es ist aber kein Nachweis dafür, dass Sie alle Anforderungen der MDR einhalten.
3.3 Ist die ISO 13485 Pflicht?
Nein, die ISO 13485 ist lediglich ein anerkannter Standard für Qualitätsmanagementsysteme für Medizinprodukte. Wer jedoch Medizinprodukte unter der MDR entwickeln möchte, ist mit einer entsprechenden Zertifizierung gut beraten. Vor allem eine benannte Stelle wird Ihr Qualitätsmanagementsystem unter die Lupe nehmen und ohne Zertifizierung könnte die Argumentation für Sie schwierig werden. Auch bei Herstellerüberwachungen von Aufsichtsbehörden ist ein ISO 13485 Zertifikat hilfreich.
3.4 Was muss ich bei Produkten mit künstlicher Intelligenz (KI) beachten?
Falls Sie künstliche Intelligenz (KI) in Ihr Produkt integrieren möchten, sollten einige weitere Aspekte bei der Umsetzung der ISO 13485 beachten. In unserem umfangreichen Leitfaden zu diesem Thema erklären wir im Detail, was für Sie bei der Zertifizierung von KI-Produkten besonders relevant ist: Zum KI-Leitfaden
3.5 Wie ist die Beziehung zwischen der ISO 13485 und der MDR?
Nach MDR müssen alle Medizinprodukt-Hersteller über ein Qualitätsmanagementsystem verfügen. Wie genau dieses auszusehen hat, spezifiziert die MDR allerdings nicht. Daher wurde die ISO 13485 zum Industriestandard in Europa, wenn es um Anforderungen an Qualitätsmanagementsysteme in der Medizinprodukt-Branche geht. Die ISO 13485 ist natürlich kein 1:1 Umsetzungsleitfaden für die MDR, denn es gibt auch Forderungen, die aus der MDR direkt hervorgehen und von der ISO 13485 nicht direkt abgedeckt werden (Beispielsweise die Ernennung einer “Person responsible for regulatory compliance – PRRC”, oder ein Prozess für die Meldung von Vorkommnissen an zuständige Behörden).
Wichtig: Die MDR spricht an keiner Stelle von der Umsetzung der ISO 13485, sondern immer von “Qualitätsmanagementsystem”. Formell gibt es also keine Pflicht, die ISO 13485 einzuhalten. Eine entsprechende Zertifizierung ist aber ein starker Nachweis für die Konformität Ihres Qualitätsmanagementsystems, weshalb wir eine solche sehr empfehlen. Die meisten MDR-Auditoren erwarten dies auch.
3.6 Kann die Herstellung von Medizinprodukten ausgelagert werden?
Ja, wenn Sie sich z. B. auf den Vertrieb und das Marketing fokussieren möchten, können Sie die Herstellung des Medizinprodukts auch an einen Dienstleister – wie QuickBird Medical – auslagern. Dadurch übernimmt der Dienstleister die Rolle des Inverkehrbringers und bringt das Medizinprodukt rechtlich auf den Markt. Somit geht die regulatorische Verantwortung auf den Dienstleister über.
Risikofreie Medizinprodukt-Zertifizierung ohne internen Aufwand für Ihr Team
Als zertifizierter Inverkehrbringer übernehmen wir für Sie die Hersteller-Verantwortung für Ihre Medizinprodukt-Software, Medical App oder DiGA. So müssen Sie kein Qualitätsmanagementsystem (QMS) bei sich aufbauen und werden als Unternehmen nicht durch die Regulatorik ausgebremst.
3.7 ISO 13485 als PDF herunterladen?
Die ISO 13485 ist kostengünstig über https://www.evs.ee/en zu erwerben und kann als PDF heruntergeladen werden (auf Englisch). Natürlich erhalten Sie die Norm auch auf zahlreichen anderen Plattformen, jedoch üblicherweise zu höheren Preisen. Die deutsche Version der ISO 13485 finden Sie unter anderem bei Beuth: https://www.beuth.de/de/norm/din-en-iso-13485/332674603
4. Weitere relevante Normen für Software-Hersteller
Als Ergänzung zur ISO 13485 müssen Sie sich als Hersteller von Software-Medizinprodukten auch mit weiteren Normen beschäftigen und diese umsetzen. Allen voran ist hier die IEC 62304 zu nennen, welche Anforderungen an die Software-Lebenszyklus-Prozesse stellt. Genaueres erfahren Sie in diesem Artikel: IEC 62304: Software-Lebenszyklus-Prozesse von Medizinprodukten
. Besonders für Hersteller von Standalone-Software lohnt sich außerdem ein Blick in die IEC 82304. Hier finden sich zwar zahlreiche Verweise auf die vorhin genannte IEC 62304, jedoch enthält sie zusätzliche Anforderungen an die Validierung von Gesundheitssoftware.
Zudem wird Ihnen auffallen, dass an einigen Stellen auf das Risikomanagement als begleitender Prozess verwiesen wird. Einen entsprechenden Prozess setzen Sie am besten nach ISO 14971 um, welcher durch die IEC 62366 (Anwendung der Gebrauchstauglichkeit auf Medizinprodukte) ergänzt wird. Weitere Informationen finden Sie in diesem Artikel: Leitfaden für die Entwicklung von Medical Apps: Darauf müssen Hersteller achten
Natürlich gibt es noch andere Normen, die für Sie relevant sein könnten. Unsere Auflistung beinhaltet lediglich jene, welche aus unserer Sicht den größten Implementierungsaufwand benötigen.
5. Medizinprodukt-Zulassung ohne Qualitätsmanagement-System?
Gibt es einen Weg, die regulatorischen Pflichten für Medizinprodukt-Hersteller komplett zu vermeiden?
Ja, den gibt es: die Auslagerung der sogenannten Legalhersteller-Rolle an ein externes Unternehmen. Dies ist eine effiziente und risikominimierende Lösung, um Ihre Ressourcen zu schonen und gleichzeitig regulatorische Sicherheit zu schaffen. Sie müssen kein Qualitätsmanagement-System aufbauen und können den regulatorischen Zulassungsprozess vollständig an uns abgeben.
So können Sie sich auf die Produktkonzeption und Vermarktung konzentrieren, während QuickBird Medical als Legalhersteller alle regulatorischen Anforderungen übernimmt und für die Einhaltung haftet.
Mehr Informationen dazu finden Sie hier: Zum Fachartikel
6. Fazit
Die ISO 13485 ist die wohl verbreitetste Norm für den Aufbau von Qualitätsmanagementsystemen für Medizinprodukt-Hersteller in Europa. Wenn Sie diese einhalten, decken Sie die meisten Anforderungen der MDR in Bezug auf Qualitätsmanagementsysteme ab.
Die Unterkapitel in den Kapiteln 4 bis 8 beschreiben jeweils einzelne Anforderungen, die von Ihnen als Hersteller umzusetzen sind und stellen somit bereits die ideale Basis für einen entsprechenden Fahrplan dar.
Auch wenn die ISO 13485 für die durchaus verständlich geschrieben ist, ist ihre Umsetzung nicht ganz selbsterklärend. Die Besonderheiten einzelner Unternehmen müssen stets berücksichtigt werden, weshalb es kein pauschales Qualitätsmanagementsystem gibt, welches alle Software-Unternehmen gleichermaßen anwenden können.
Dennoch ist es ratsam, sich entsprechende Vorlagen zu kaufen und diese anschließend anzupassen.
Zudem empfehlen wir, den Aufbau Ihres Qualitätsmanagementsystems gemeinsam mit einem erfahrenen Berater vorzunehmen. Entsprechende Software-Tools können Ihnen außerdem einen maßgeblichen Effizienz-Vorteil verschaffen.
Dennoch, der Aufbau eines Qualitätsmanagementsystems ist sehr zeitaufwändig und produziert Kosten. Daher kann es für Sie sinnvoll sein, die Rolle des Inverkehrbringers für Ihr Produkt (zunächst) an einen Dienstleister zu übergeben. Dieser übernimmt dann die regulatorische Verantwortung für Ihr Produkt und steht rechtlich für seine Konformität ein. In diesem Fall entgehen Sie der Pflicht, ein eigenes Qualitätsmanagementsystem aufzubauen.
Wenn Sie die Umsetzung eines Software-Medizinprodukts und/oder den Aufbau eines Qualitätsmanagementsystems planen, sprechen Sie uns gerne an. Wir unterstützen Sie bei Ihrem Vorhaben. Kontaktieren Sie uns jetzt.
The post ISO 13485 – Leitfaden zu Anforderungen und Inhalten appeared first on QuickBird Medical.
]]>The post Datensicherheits- & Datenschutz-Zertifikate für DiGA appeared first on QuickBird Medical.
]]>- Datensicherheits-Zertifikat nach BSI TR-03161
- Datenschutz-Zertifikat nach DSGVO
Hinzu kommt das Vorhalten eines nach ISO/IEC 27001 zertifizierten Informationssicherheitsmanagementsystems, welches jedoch nicht im Fokus dieses Artikels steht.
Wir erklären, was es mit dem Datenschutz- und Datensicherheits-Zertifikat auf sich hat, ab wann die Zertifizierungen verpflichtend sind und wie Sie diese als Hersteller erhalten können.
Inhaltsverzeichnis
- 1. Ab wann gilt die Pflicht zur Zertifizierung
- 2. Wie bekomme ich das DiGA-Zertifikat für Datensicherheit?
- 3. Wie bekomme ich das DiGA-Zertifikat für Datenschutz
- 4. Fazit
- 5. Vorbereitung auf die Zertifizierung
1. Ab wann gilt die Pflicht zur Zertifizierung?
Die aktuellen Fristen für die Zertifizierung von DiGA nach Datenschutz und Datensicherheit lauten wie folgt:
Datensicherheitszertifikat nach BSI TR-03161: 01.01.2025
Seit dem 1. Januar 2025 sind Hersteller digitaler Gesundheitsanwendungen (DiGA) verpflichtet, die Einhaltung der Anforderungen an die Datensicherheit durch ein offizielles Zertifikat zu belegen.
- Neue DiGA-Anträge: Für neue DiGA-Anträge gab es zwischenzeitlich eine Übergangsregelung, die nun aber nicht mehr relevant ist. Um in das DiGA-Verzeichnis aufgenommen zu werden, benötigen Sie aktuell zwingend eine Datensicherheitszertifizierung nach BSI TR-03161.
- Gelistete DiGA: Für bereits gelistete DiGA duldet das BfArM aktuell noch fehlende Zertifikate. Voraussetzung ist, dass der Hersteller nachweisen kann, dass er sich im laufenden Zertifizierungsprozess befindet. Diese Praxis kann sich jederzeit ändern. Hersteller gelisteter DiGA sollten sicherstellen, dass sie das Zertifikat des BSI sehr zeitnah erhalten.
Datenschutzzertifikat nach DSGVO: weiterhin unklar
Aktuell besteht für DiGA noch keine Pflicht zur Vorlage eines formalen Datenschutzzertifikats. Der Grund dafür ist, dass es zum jetzigen Zeitpunkt noch keine akkreditierten Zertifizierungsstellen für eine Zertifizierung nach den Datenschutzkriterien gemäß § 139e Absatz 11 SGB V und § 78a Absatz 8 SGB XI gibt.
Das BfArM arbeitet derzeit gemeinsam mit dem BfDI und dem BSI an der Umsetzung der gesetzlichen Vorgaben. Die zugrunde liegenden Datenschutzkriterien wurden bereits aktualisiert und veröffentlicht. Sie bilden künftig die Basis für ein offizielles Datenschutzzertifikat, das sowohl die Anforderungen der DSGVO als auch zusätzliche DiGA- und DiPA-spezifische Vorgaben abdeckt. Das Zertifikat befindet sich aber weiterhin in der Entwicklung. Änderungen an Prüfkriterien und Prüfmethoden sind daher möglich.
Solange eine Zertifizierung technisch und organisatorisch noch nicht möglich ist, gelten weiterhin die Datenschutzanforderungen der DiGAV.
Sobald Zertifizierungsstellen akkreditiert sind, wird das BfArM die Vorlage eines Datenschutzzertifikats mit ausreichendem zeitlichem Vorlauf einfordern. Konkrete Zeiträume werden auf der Webseite des BfArM veröffentlicht. Hersteller bereits gelisteter DiGA werden dann individuell zur Vorlage des Zertifikats aufgefordert.
2. Wie bekomme ich das DiGA-Zertifikat für Datensicherheit?
Das Datensicherheitszertifikat für DiGA basiert auf der BSI TR-03161 und wird über ein mehrstufiges Verfahren vergeben. Voraussetzung ist eine vollständige technische und organisatorische Umsetzung der Anforderungen.
Der Weg zum Zertifikat in Kurzform:
- Vorbereitung: Umsetzung der technischen und organisatorischen Anforderungen der BSI TR-03161 im Produkt und in den Prozessen des Herstellers.
- Prüfung durch eine akkreditierte Prüfstelle: Eine vom BSI zugelassene Prüfstelle prüft Produkt, Architektur, Quellcode, Dokumentation und Sicherheitsprozesse und erstellt einen Prüfbericht.
- Zertifizierungsentscheidung durch das BSI: Auf Basis des Prüfberichts entscheidet das BSI über die Erteilung des Zertifikats und stellt dieses bei positiver Bewertung aus.
Wir haben einen detaillierten Leitfaden geschrieben, der Ihnen einen Überblick über den gesamten Prozess der Datensicherheitszertifizierung nach BSI TR-03161 gibt. Wir befassen uns in diesem Rahmen damit:
- Was fordert die TR-03161 konkret?
- Wie läuft die Zertifizierung mit Prüfstelle und BSI ab?
- Mit welchen Kostenblöcken ist das Ganze verbunden und wie lange dauert eine solche Zertifizierung?
- Was sind die praktischen Implikationen der BSI TR-03161 auf DiGA?
- Wie sieht die Zukunft der BSI-Zertifizierung aus? (Stichwort: BSI TR-03185)
Hier finden Sie unseren Leitfaden zur BSI TR-03161-Zertifizierung: Zum Fachartikel
3. Wie bekomme ich das DiGA-Zertifikat für Datenschutz?
Das Datenschutz-Zertifikat ist eine Zertifizierung nach Artikel 42 DSGVO. Die zugrundeliegende Prüfung soll anhand von Prüfkriterien erfolgen, die vor einigen Monaten vom BfArM veröffentlicht wurden: DiGA und DiPA Datenschutzkriterien
Entsprechende Zertifikate können laut DSGVO von den Aufsichtsbehörden selbst, oder aber von akkreditierten Stellen ausgestellt werden. In der Praxis zeigt sich hier aber noch ein anderes Bild.
Das Bayerische Bundesamt für Datenschutzaufsicht (BayLDA) zum Beispiel führt „mangels personeller Ressourcen“ keine Zertifizierungen selbst durch. Stattdessen wird auf die bei der DAkkS (Deutsche Akkreditierungsstelle) akkreditierten Organisationen verwiesen, welche es aktuell aber auch noch nicht zu geben scheint (Quelle: BayLDA – Zertifizierung). (Stand vom 15.03.2024)
Viele Fragen, die DiGA-Hersteller beschäftigen, können heute leider noch nicht geklärt werden. Dazu zählen unter anderem:
- Wer kann entsprechende Prüfungen durchführen, die für die Zertifizierung benötigt werden?
- Wie lange dauern die Zertifizierungsverfahren?
- Wie viel kosten die Zertifizierungen?
- etc.
Die zentralen Informationen fehlen also noch, mit denen DiGA-Hersteller die gesetzlichen Vorgaben erfüllen können. Aktuell ist somit noch keine Datenschutzzertifizierung möglich.
Sobald sich dies ändert oder Neuigkeiten bekannt werden, benachrichtigen wir alle Abonnenten unseres DiGA-Newsletters. Wenn Sie hierzu auf dem neuesten Stand bleiben möchten, abonnieren Sie unseren Newsletter für DiGA-Hersteller hier.
4. Fazit
Wir beobachten die Situation mit der Datenschutzzertifizierung für alle unsere DiGA-Kunden aktiv und sind gespannt, wann es hierzu Neuigkeiten geben wird. Fakt ist: Die Datenschutzzertifizierung wird kommen. Es ist nur eine Frage der Zeit, wann dies so weit ist.
In Bezug auf die Datensicherheitszertifizierung existiert bereits ein Prüfprozess und über 10 DiGA sind bereits nach BSI TR-03161 zertifiziert (Stand Januar 2026). Der Prüfprozess hat an vielen Stellen Verbesserungspotenzial und daher ist auch hier zeitnah mit Updates zu rechnen. Mehr dazu finden Sie in unserem Fachartikel zur BSI-Zertifizierung von DiGA.
The post Datensicherheits- & Datenschutz-Zertifikate für DiGA appeared first on QuickBird Medical.
]]>The post Ist Ihre App eine DiGA? Definition & Kriterien digitaler Gesundheitsanwendungen appeared first on QuickBird Medical.
]]>Update des DiGA-Leitfadens am 10.12.2025: Der offizielle DiGA-Leitfaden wurde in der Version 3.6 aktualisiert und enthält zahlreiche Ergänzungen in Bezug auf die Entscheidung „Ist Ihre Anwendung eine DiGA?“ (Qualifizierung). Die Neuerungen wurden in den untenliegenden Fachartikel bereits eingearbeitet. Einen Überblick über die Leitfaden-Änderungen finden Sie hier: Übersicht aller Änderungen
Inhaltsverzeichnis
- 1. DiGA-Kriterien im Überblick
- 1.1 Kriterium #1: Medizinprodukt niedriger oder höherer Risikoklasse
- 1.2 Kriterium #2: Positiver Versorgungseffekt, konsistent mit Zweckbestimmung des Medizinprodukts
- 1.3 Kriterium #3: Digitale Hauptfunktion
- 1.4 Kriterium #4: Mehr als eine Steuerung von Geräten
- 1.5 Kriterium #5: Diagnose oder Therapie von Krankheiten oder Verletzungen
- 1.6 Kriterium #6: Keine reine (Primär-)Prävention
- 1.7 Kriterium #7: Nutzer ist der Patient
- 1.8 Kriterium #8: Individualisierung
- 1.9 Kriterium #9: Ausgeschlossene Leistungen
- 1.10 Kriterium #10: Veröffentlichung vor Antrag
- 2. Der DiGA-Fähigkeit-Entscheidungsbaum
- 3. Die Anforderungen bei Erfüllung der Kriterien
- 4. Selektivverträge – Eine Alternative zu DiGA
- 5. Fazit
1. DiGA-Kriterien im Überblick
Der offizielle DiGA-Leitfaden des BfArM fasst die Definition von DiGA prägnant zusammen. Damit Ihr Produkt als DiGA für eine Kostenerstattung potenziell in Frage kommt, muss es die folgenden Kriterien erfüllen.
1.1 Kriterium #1: Medizinprodukt niedriger oder höherer Risikoklasse
Ihr Produkt muss ein Medizinprodukt niedriger oder höherer Risikoklasse sein (I, IIa oder IIb).
Natürlich ist nicht jede Software ein Medizinprodukt. Eine App, die beispielsweise die reine Dokumentation von medizinischen Daten zum Zweck hat, ist kein Medizinprodukt und damit auch keine DiGA. Falls Ihre App Blutdruck-Werte sammelt, diese jedoch nicht für die Diagnose oder Therapie von Krankheiten auswertet, ist dies kein Medizinprodukt. Mehr Informationen finden Sie in unserem Artikel zum Thema „Ist Ihre App ein Medizinprodukt?“.
Ihre App ist ein Medizinprodukt? Super! Allerdings ist dann aber noch offen, ob sie auch ein Medizinprodukt einer niedrigen oder höheren Risikoklasse (I, IIa oder IIb) ist.
Zur Bestimmung der Risikoklasse ist insbesondere die Regel 11 der MDR relevant. Auch hierzu haben wir einen detaillierten Leitfaden geschrieben: Praxis-Leitfaden zur Bestimmung der Risikoklasse bei Software-Produkten
1.2 Kriterium #2: Positiver Versorgungseffekt, konsistent mit Zweckbestimmung des Medizinprodukts
Die digitale Hauptfunktion der DiGA muss konsistent mit der Zweckbestimmung des zugrundeliegenden Medizinproduktes sein. Funktionen, die Teil der Erfüllung der medizinischen Zweckbestimmung sind, müssen auch den Funktionsumfang der DiGA ausmachen.
Hierbei sind u.a. folgende Aspekte relevant:
- Exkludieren von DiGA-Funktionen: Manchmal hat eine DiGA bestimmte Funktionen, die man als Hersteller lieber nicht den strikten Anforderungen der DiGA-Verordnung unterwerfen will. Manche Funktionen erschweren oder verhindern z.B. die BSI-Sicherheitszertifzierung der DiGA oder den Nachweis des positiven Versorgungseffekts. Dennoch gilt: Wenn diese Funktionen zur Erfüllung der Zweckbestimmung des Medizinprodukts dienen, dürfen Sie diese nicht vom Funktionsumfang der DiGA exkludieren. Sie dürfen also nicht beliebige Funktionen aus dem Scope Ihrer DiGA ausschließen, auch wenn dies die initiale DiGA-Zulassung für Sie erschwert oder verhindert.
- Inkonsistenz mit Zweckbestimmung: Eine andere Form der Inkonsistenz zwischen Medizinprodukt-Zweckbestimmung und Hauptfunktion der DiGA wäre Folgendes: Ihre DiGA zielt auf eine andere medizinische Indikation ab als das zugrunde liegende Medizinprodukt. Ihr Medizinprodukt dient der Therapie von Kniegelenksarthrose, aber Ihre DiGA hat die Handgelenksarthrose als Hauptfunktion. Das wäre eine Inkonsistenz in diesem Sinne und würde sich so nicht als DiGA qualifizieren.
1.3 Kriterium #3: Digitale Hauptfunktion
Ihr Produkt muss definitionsgemäß hauptsächlich auf digitalen Technologien beruhen. Das schließt aus, dass z.B. ein Blutdruckmessgerät selbst eine DiGA ist. Hardware ist keine DiGA.
Aber auch wenn Ihr Produkt z.B. ein App oder Web-Anwendung ist und somit scheinbar vollständig digital, sollten Sie beachten:
- Hardware-Integration: Wenn Sie eine Hardware z.B. per Bluetooth mit Ihrer App verbinden: Der positive Versorgungseffekt Ihrer DiGA darf dann nicht durch den Einsatz der Hardware entstehen. Der positive Versorgungseffekt muss auf der digitalen Hauptfunktion (z.B. digitales Übungsprogramm) basieren und somit unabhängig von der Hardware sein.
- Einbindung von Behandlern: Der positive Versorgungseffekt darf nicht durch die Einbindung z.B. eines Arztes entstehen. Wenn Ihre DiGA nur „wirkt“, wenn ein Arzt über Video-Calls persönliche Therapieempfehlungen gibt, dann ist das keine digitale Hauptfunktion Ihrer DiGA. Die DiGA darf für den positiven Versorgungseffekt nicht von dieser menschlichen Komponente abhängen. Diese muss optionalen Charakter haben, und im Optimalfall bei der Durchführung der DiGA-Studie in der Interventionsgruppe deaktiviert werden.
1.4 Kriterium #4: Mehr als eine Steuerung von Geräten
Ihre App muss den medizinischen Zweck wesentlich durch die digitale Hauptfunktion erfüllen. Falls Ihre App nur der Steuerung eines medizinischen Geräts dient – und damit den wesentlichen medizinischen Zweck nicht selbst erbringt – ist sie keine DiGA. Eine DiGA kann zwar mit medizinischen Geräten kommunizieren und z.B. Sensordaten auslesen, dies darf jedoch nicht der Hauptzweck sein.
Falls Ihre App externe Sensordaten eines medizinischen Geräts ausliest und auf dieser Basis eine Diagnose stellt oder Therapieempfehlungen gibt, ist dieses Kriterium erfüllt. Der wesentliche Zweck ist in diesem Fall Diagnose oder Therapieverbesserung, und der Zweck wird durch einen digitalen Algorithmus in Ihrer App/Software erfüllt.
1.5 Kriterium #5: Diagnose oder Therapie von Krankheiten oder Verletzungen
Eine DiGA wird definiert als eine Anwendung, welche die „Erkennung, Überwachung, Behandlung oder Linderung von Krankheiten oder die Erkennung, Behandlung, Linderung oder Kompensierung von Verletzungen oder Behinderungen“ unterstützt. Damit sind z.B. Verhütungs-Apps, Zyklus-Tracking-Apps, oder reine (Primär-)Präventions-Apps (Kriterium #5) keine DiGA. Eine Applikation, die beispielsweise Patienten mit diagnostizierter Sehschwäche durch digitale Sehübungen bei der Therapie unterstützt, erfüllt dieses Kriterium und könnte demnach auch eine DiGA sein.
1.6 Kriterium #6: Keine reine (Primär-)Prävention
Der DiGA-Leitfaden formuliert es folgendermaßen: Voraussetzung ist, dass ein Risikofaktor im Sinne einer Erkrankung vorliegt, die als Diagnose verschlüsselbar ist.
Eine App, die einen gesunden Menschen bei der Prävention von Krankheiten unterstützt, ist keine DiGA. Auch (Primär-)Präventions-Apps sind potentiell Medizinprodukte, jedoch damit noch keine DiGA. Denn eine DiGA wird z.B. vom Arzt im Falle einer Krankheits-Diagnose verschrieben und richtet sich damit immer an Personen mit einer bestimmten Indikation (ICD-10 Code).
1.7 Kriterium #7: Nutzer ist der Patient
Eine App, die hauptsächlich den Arzt bei seiner täglichen Arbeit unterstützt, ist keine DiGA. Eine DiGA muss für die Nutzung durch den Patienten bestimmt sein. Dabei kann die App durchaus den Arzt mit einbeziehen, solange der Patient der Hauptnutzer bleibt.
1.8 Kriterium #8: Individualisierung
DiGA müssen auf Basis der Eingaben der Patienten individualisierte Rückmeldungen generieren.
Beispiel: Ihre DiGA frägt in einem Onboarding-Fragebogen den Gesundheitszustand des Patienten ab. Danach erhält der Patient eine gleichbleibende Auswahl von Wissensinhalten und Übungen.
Das würde höchstwahrscheinlich das Individualisierungskriterium nicht erfüllen. Die Wissensinhalte und Übungen sollten stattdessen auf Basis der eingegebenen Werte aufbereitet werden. Sie könnten z.B. Patienten mit verschiedenem Schwergrad oder Stadium der Krankheit auch verschiedene Wissensinhalte und Übungen anzeigen. So individualisieren Sie die DiGA auf Basis der eingegebenen Daten.
1.9 Kriterium #9: Ausgeschlossene Leistungen
Die DiGA umfasst keine Leistungen, die nach dem Dritten Kapitel des SGB V ausgeschlossen sind. Ebenso darf sie keine Inhalte enthalten, zu denen der Gemeinsame Bundesausschuss bereits eine ablehnende Entscheidung nach den §§ 92, 135 oder 137c getroffen hat.
1.10 Kriterium #10: Veröffentlichung vor Antrag
Zum Zeitpunkt der Antragstellung muss die DiGA bereits verfügbar sein. Sie muss in den entsprechenden App Stores veröffentlicht sein oder im Fall einer Webanwendung über die vorgesehene Website öffentlich zugänglich sein.
2. Der DiGA-Fähigkeit-Entscheidungsbaum
Im DiGA-Leitfaden der Version 3.6 hat das BfArM eine übersichtliche Grafik eingefügt, die Ihnen bei der Qualifizierung Ihrer Anwendung als DiGA helfen soll:

Abbildung 3 aus dem DiGA-Leitfaden: DiGA-Fähigkeit Entscheidungsbaum. Quelle: BfArM.
Weitere Informationen zu allen Kriterien und Anforderungen finden Sie im offiziellen DiGA-Leitfaden des BfArM.
3. Die Anforderungen bei Erfüllung der Kriterien
Natürlich wird Ihre Software bei Erfüllung dieser Kriterien nicht automatisch in das DiGA-Verzeichnis aufgenommen und erstattet. Falls Sie grundsätzlich jedes dieser Kriterien erfüllen, müssen Sie sich im nächsten Schritt mit den Anforderungen an DiGA befassen. Diese Anforderungen regelt die Digitale Gesundheitsanwendungen-Verordnung (DiGAV). Im offiziellen DiGA-Leitfaden werden diese dann weiter ausgeführt.
Zu den Anforderungen gehören zum einen die „Anforderungen an Sicherheit, Funktionstauglichkeit, Datenschutz und -sicherheit sowie Qualität digitaler Gesundheitsanwendungen“. Zusammengefasst umfasst dieser Punkt folgende Anforderungen:
- Anforderungen an Sicherheit und Funktionstauglichkeit
- Anforderungen an Datenschutz und Datensicherheit
- Anforderungen an Qualität und Interoperabilität
- Anforderungen an Robustheit
- Anforderungen an Verbraucherschutz
- Anforderungen an Nutzerfreundlichkeit
- Anforderungen an die Unterstützung der Leistungserbringer
- Anforderungen an die Qualität der medizinischen Inhalte
- Anforderungen an die Patientensicherheit
Außerdem müssen Sie mit Ihrer DiGA einen positiven Versorgungseffekt nachweisen. Ein positiver Versorgungseffekt ist laut DVG entweder ein medizinischer Nutzen oder patientenrelevante Struktur- und Verfahrensverbesserungen in der Versorgung. Diesen positiven Versorgungseffekt müssen Sie innerhalb wissenschaftlicher Studien belegen. Falls Sie derartige Studien zum Antragszeitpunkt nicht vorliegen haben, müssen Sie zumindest ein wissenschaftliches Evaluationskonzept vorlegen, mit dem Sie zu einem späteren Zeitpunkt die Versorgungseffekte nachweisen möchten.
Des Weiteren müssen Sie z.B. ein Datensicherheitszertifizierung beim BSI erlangen und Ihre DiGA an die elektronische Patientenakte anbinden.
4. Selektivverträge – Eine Alternative zu DiGA
Sollte sich Ihre digitale Lösung nicht als DiGA qualifizieren, gibt es in Deutschland auch die Möglichkeit, Selektivverträge mit einzelnen Krankenkassen abzuschließen. Dies ermöglicht Ihnen einen Weg in die Erstattung, der auf Ihre Anwendung zugeschnitten ist.
In unserem Leitfaden zu Selektivverträgen geben wir Ihnen einen Überblick über diese alternative Form der Kostenerstattung: Zum Leitfaden – Selektivverträge für digitale Lösungen
Sogar über Selektivverträge und DiGA hinaus gibt es noch weitere Wege in die Erstattung für Software-Produkte:
- Überblick über alle Wege in die Kostenerstattung: Zu unserem Whitepaper
- Erstattung von Software im GKV-Hilfsmittelverzeichnis (HMV): Zu unserem Leitfaden
- Digitale Pflegeanwendungen – DiPA-Verfahren: Zu unserem Leitfaden
- Zentrale Prüfstelle Prävention (ZPP): Zu unserem Leitfaden
Neben den genannten Wegen in die Erstattung lohnt auch der Blick auf öffentliche Fördermöglichkeiten, die den Weg dahin überbrücken können. Allen voran lohnt sich ein Blick auf den Innovationsfonds des G-BA, der digitale Versorgungslösungen in frühen Phasen finanziell unterstützt.
Mehr Informationen dazu finden Sie in unserem Leitfaden zum Innovationsfonds: Zum Leitfaden
5. Fazit
Ob Ihre App potenziell eine DiGA ist, wird durch die genannten Kriterien definiert. Der Teufel steckt allerdings oft im Detail. Sie sollten sich also für die Planung auch den offiziellen DiGA-Leitfaden genau durchlesen und von Erfahrungen anderer DiGA-Hersteller lernen. Auch wenn der DiGA-Leitfaden umfangreich ist und sicherlich viele Hilfestellungen bietet, gibt es leider weiterhin viel implizites Wissen bei den existierenden DiGA-Herstellern, welches dort noch nicht abgebildet wurde.
QuickBird Medical entwickelt DiGA auf Auftragsbasis, seit es sie gibt. Stand heute (2025) haben wir bereits über 15 DiGA-Projekte umgesetzt. Falls Sie eine DiGA planen und die Entwicklung sowie die Regulatorik an einen Dienstleister auslagern möchten, kontaktieren Sie uns gerne.
The post Ist Ihre App eine DiGA? Definition & Kriterien digitaler Gesundheitsanwendungen appeared first on QuickBird Medical.
]]>The post BSI TR-03161 für DiGA: Zertifizierung der Datensicherheit appeared first on QuickBird Medical.
]]>Für die Listung einer Anwendung im DiGA-Verzeichnis prüft das BfArM die Zertifizierung als feste Voraussetzung. Ohne gültiges Zertifikat werden keine neuen DiGA mehr aufgenommen. Und auch gelisteten DiGA droht ohne Zertifizierung eine Streichung aus dem Verzeichnis (der Stichtag für gelistete DiGA existiert allerdings aktuell nicht).
Viele Hersteller unterschätzen, wie umfangreich die Implikationen dieser Zertifizierung sind und wie aufwendig sich der Prüfprozess gestaltet.
Mit diesem Artikel möchten wir (angehenden) DiGA-Herstellern einen strukturierten Überblick geben:
- Was fordert die TR-03161 konkret?
- Wie läuft die Zertifizierung mit Prüfstelle und BSI ab?
- Mit welchen Kostenblöcken ist das Ganze verbunden und wie lange dauert eine solche Zertifizierung?
- Was sind die praktischen Implikationen der BSI TR-03161 auf DiGA?
- Wie sieht die Zukunft der BSI-Zertifizierung aus? (Stichwort: BSI TR-03185)
Auf Basis unserer praktischen Erfahrungen aus mehreren durchgeführten TR-03161-Projekten schildern wir den aktuellen Stand der Zertifizierungsanforderungen.
Inhaltsverzeichnis
- 1. Für wen gilt die BSI TR-03161?
- 2. Anforderungen und Aufbau der BSI TR-03161
- 3. Der Zertifizierungsprozess
- 4. Kosten und Zeitrahmen des Zertifizierungsprozesses
- 5. Gültigkeitsdauer & Rezertifizierung nach BSI TR-03161
- 6. BSI TR-03161-Zertifikat mit Auflagen
- 7. Änderungen an zertifizierten Produkten – BSI TR-03185
- 8. Praktische Implikationen für DiGA
- 9. Verpflichtender Penetrationstest für DiGA
- 10. BSI TR-03161 vs. ISO 27001
- 11. Kritik am Prozess
- 12. Wichtige Links und Ressourcen
- 13. Zukunftsausblick für die BSI TR-03161 und DiGA-Datensicherheitszertifizierung
- 14. Fazit
1. Für wen gilt die BSI TR-03161?
Die TR-03161 kann grundsätzlich für viele Anwendungen im deutschen Gesundheitswesen genutzt werden, die sensible Daten verarbeiten.
Gesetzlich verpflichtend ist sie jedoch vor allem für digitale Gesundheitsanwendungen (DiGA) nach § 139e SGB V und digitale Pflegeanwendungen (DiPA) nach § 78a SGB XI. Für diese Produkte muss ab 1. Januar 2025 ein offizielles Zertifikat vorgelegt werden.
In diesem Artikel liegt der Fokus auf DiGA, da hier die Zertifizierungspflicht aktuell die größte praktische Relevanz hat.
Für die Zertifizierung nach BSI TR-03161 ist zu beachten:
- Sie ist unabhängig von der Risikoklasse anwendbar
- Sie umfasst sämtliche Komponenten der Anwendung (also App, Backend, Webportal sowie Patienten- und Behandlerzugänge)
- Auch wenn Hosting oder Infrastruktur an externe Cloud-Anbieter ausgelagert werden, müssen diese Komponenten sich an die Anforderungen der BSI TR-03161 halten. Gleichzeitig müssen die Server-Anbieter ein C5-Testat vom Typ 2 vorweisen.
- Neue DiGA benötigen für die Aufnahme in das DiGA-Verzeichnis bereits jetzt eine Zertifizierung nach BSI TR-03161.
- Für zum aktuellen Zeitpunkt bereits gelistete DiGA wird es zurzeit vom BfArM noch geduldet, dass kein Zertifikat vorliegt, solange der Hersteller nachweisen kann, dass er sich im Zertifizierungsprozess befindet. Das kann sich allerdings jederzeit ändern.
Hinweis: Wir bezeichnen das Produkt eines (angehenden) DiGA-Herstellers in diesem Artikel einfachheitshalber immer als „DiGA“, unabhängig davon, ob diese bereits gelistet ist oder nicht. Natürlich ist aber nicht jede Anwendung bereits als DiGA gelistet, sobald sie den Zertifizierungsprozess nach BSI TR-03161 beginnt.
2. Anforderungen und Aufbau der BSI TR-03161
Die Technische Richtlinie TR-03161 definiert die Mindestanforderungen an Datensicherheit für digitale Gesundheits- und Pflegeanwendungen. Damit bildet sie die Grundlage für die gesetzlich geforderte Datensicherheitszertifizierung nach § 139e SGB V und § 78a SGB XI.
IT-Sicherheit im Allgemeinen und auch die BSI TR-03161 verfolgen im Wesentlichen drei Schutzziele: Vertraulichkeit, Integrität und Verfügbarkeit. Die Norm legt durch konkrete Maßnahmen fest, wie Sie als Hersteller sicherstellen müssen, dass sensible Daten vertraulich, integer und verfügbar bleiben.
- Vertraulichkeit bedeutet, dass nur berechtigte Personen oder Systeme auf sensible Daten zugreifen dürfen. Unbefugte dürfen Daten nicht einsehen oder mitlesen, weder in der App noch bei der Übertragung.
- Integrität stellt sicher, dass Daten korrekt und unverändert bleiben. Manipulationen oder unbeabsichtigte Änderungen werden verhindert oder eindeutig erkennbar gemacht.
- Verfügbarkeit bedeutet, dass Anwendung und Daten zuverlässig nutzbar sind. Ausfälle oder Unterbrechungen werden durch geeignete technische und organisatorische Maßnahmen minimiert.
2.1 Aufbau der BSI TR-03161
Die TR-03161 besteht aus drei Teilen, die zusammen alle sicherheitsrelevanten Komponenten einer digitalen Gesundheitsanwendung abdecken. Jeder Teil fokussiert sich auf eine bestimmte Systemebene und formuliert spezifische Anforderungen, die später im Zertifizierungsaudit einzeln geprüft werden. So entsteht ein vollständiges Sicherheitsmodell, das App, Web-Frontend und Backend gleichermaßen absichert.
Die drei Teile der Richtlinie decken alle technischen Komponenten einer DiGA ab. App, Web-Frontend und Backend werden jeweils separat geprüft und zertifiziert.
- Teil 1: Mobile Anwendungen (Apps): Teil 1 beschreibt die Sicherheitsanforderungen an mobile Anwendungen (beispielsweise iOS und Android). Er legt fest, wie Apps sensible Daten verarbeiten dürfen, wie sie Gerätefunktionen nutzen und wie sie vor Manipulation geschützt werden müssen.
- Teil 2: Web-Anwendungen: Teil 2 richtet sich an webbasierte Komponenten (z. B. eine Web-Oberfläche, die ein Behandler oder Patient im Browser aufrufen kann). Der Teil beschreibt, wie Webanwendungen im Browser sicher ausgeführt werden und wie typische Web-Schwachstellen verhindert werden müssen.
- Teil 3: Hintergrundsysteme (Backend): Teil 3 definiert die Anforderungen an serverseitige Komponenten, unabhängig davon, ob diese On-Premise oder in der Cloud betrieben werden. Da sensible Gesundheitsdaten in der Regel zentral verarbeitet werden, enthält dieser Teil besonders umfangreiche Vorgaben zur Architektur und zum Betrieb.
Wichtig ist hier, dass für jede DiGA individuell entschieden wird, welche dieser 3 Teile für das Produkt zutreffen. Eine Web-Anwendung mit Backend-Server muss z. B. nur Teil 2 und 3 und nicht Teil 1 (für mobile Anwendungen) im Rahmen der Zertifizierung berücksichtigen.
2.2 Umfang: Sowohl Produkt als auch Prozesse
Der Aufbau der Richtlinie folgt einer klaren Systematik, die auf einer sogenannten Security Problem Definition basiert. Diese umfasst:
- Annahmen über Browser, Endgerät und Backend-Infrastruktur
- definierte Bedrohungsszenarien wie unbefugten Zugriff, Datenmanipulation, Ausnutzen von Debug-Funktionen oder Fehlkonfigurationen
- organisatorische Sicherheitsrichtlinien, die der Hersteller umsetzen muss, zum Beispiel: Security-Lifecycle, Patch-Management, Offenlegung des Datenverarbeitungszwecks, Prozesse zur Meldung von Schwachstellen
Die TR-03161 betrachtet damit nicht nur die technische Umsetzung (Quellcode und Produkt), sondern auch Prozesse und Dokumentation. Ein produktbezogener Pen-Test reicht also nicht für eine Zertifizierung. Auch die Prozesse und die Dokumentation des Herstellers werden überprüft.
2.3 Prüfaspekte
Die Richtlinie strukturiert alle Anforderungen in Prüfaspekte. Jeder Prüfaspekt enthält konkrete Regeln, die mit MUSS, SOLL, KANN oder DARF NICHT versehen sind.
Jeder Teil der BSI TR-03161 enthält 11 (bei Teil 1 und 2) bzw. 10 (bei Teil 3) Prüfaspekte:
- Anwendungszweck
- Architektur
- Quellcode
- Drittanbieter-Software
- Kryptographische Umsetzung
- Authentisierung und Authentifizierung
- Datensicherheit
- Kostenpflichtige Ressourcen
- Netzwerkkommunikation
- Organisatorische Sicherheit (nur bei Teil 3 – Hintergrundsysteme)
- Plattformspezifische Interaktionen (nur bei Teil 1 und 2 – Mobile & Web)
- Resilienz (nur bei Teil 1 und 2 – Mobile & Web)
Tipp: Kopieren Sie sich eine Liste der einzelnen Prüfaspekte jedes Teils der TR-03161 in Ihre Unterlagen und haken Sie diese strukturiert nacheinander ab. Das ist ein hilfreiches Projektmanagementinstrument für Ihr Entwicklerteam.
3. Der Zertifizierungsprozess
Der gesamte Zertifizierungsprozess für eine DiGA verläuft über drei Phasen hinweg:
- Vorbereitungsphase: Sie als DiGA-Hersteller bereiten sich auf den Prüfprozess vor. Sie arbeiten sich in die Norm BSI TR-03161 ein und setzen die Vorgaben nach bestem Gewissen innerhalb Ihres Produkts und Ihrer Prozesse um. Sie suchen sich außerdem eine Prüfstelle für die Zertifizierung, schaffen die vertragliche Basis für die Zusammenarbeit mit dieser und reservieren sich einen Prüftermin.
- Prüfphase: Sobald Sie von Ihrer Seite mit der Umsetzung der TR-Vorgaben fertig sind, schicken Sie Ihre Unterlagen, Produktzugänge und Software-Quellcode an die Prüfstelle. Diese beginnt den Prüfprozess und schickt Ihnen nach Abschluss der Prüfung den Prüfbericht inklusive möglicher Abweichungen. Ob es auch zwischenzeitliches Feedback oder andere Kommunikation gibt, hängt dabei von der jeweiligen Prüfstelle ab. Falls die Prüfstelle in allen Punkten zufrieden ist, bereitet sie den Prüfbericht für die Versendung an das BSI vor.
- Zertifizierungsphase: Die Zertifizierungsstelle des BSI erhält den fertigen Prüfbericht von der Prüfstelle und evaluiert, ob sie die Zertifizierung auf dieser Basis positiv bewerten kann. Wenn ja, erhalten Sie vom BSI nun das Zertifikat inklusive Konformitätsreport. Wenn nicht, schickt das BSI eine kommentierte Liste des Prüfberichts an die Prüfstelle zurück. Diese evaluiert das Feedback und gibt die für Sie als Hersteller relevanten Abweichungen an den DiGA-Hersteller zur Umsetzung weiter.
Hinweis: Die Prüfstelle bezeichnet ein privates Unternehmen, das durch das BSI offiziell für die Prüfung dieser technischen Richtlinie akkreditiert wurde. Eine Liste der Prüfstellen finden Sie hier unten im Artikel. Mit Zertifizierungsstelle bezieht man sich hingegen auf die Abteilung beim BSI, die für die Zertifizierung zuständig ist und darüber in letzter Instanz entscheidet.
Den Ablauf des Zertifizierungsprozesses haben auch wir in vereinfachter Form in der folgenden Abbildung skizziert.

BSI-Zertifizierungsprozess für DiGA-Hersteller nach TR-03161
4. Kosten und Zeitrahmen des Zertifizierungsprozesses
4.1 Kosten des Zertifizierungsprozesses
Für die interne Budgetplanung sollten Sie mit folgenden Kostenpaketen rechnen:
- Die Kosten der Prüfstelle: Für die Arbeit der Prüfstelle zur Zertifizierung fallen natürlich Kosten an. Die Höhe dieser Kosten variiert zwischen den Prüfstellen und hängt außerdem von Ihrem Produkt und dem Umfang der Prüfung ab. Wir empfehlen Ihnen, mehrere Angebote einzuholen, um diese zu vergleichen.
- Die Kosten für das BSI: Gegebenenfalls weniger offensichtlich ist, dass auch das BSI nach Abschluss des Zertifizierungsprozesses eine Rechnung an Sie schicken wird. Das passiert übrigens unabhängig davon, ob die Prüfung erfolgreich war oder nicht. Die Gebührenordnung finden Sie hier.
- Entwicklungskosten: Für das Entwicklerteam werden signifikante Aufwände bei der Implementierung der TR-Anforderungen anfallen. Die damit zusammenhängenden Kosten sollten Sie natürlich auch einplanen.
- Beratungskosten: Es empfiehlt sich, mit einem Team zusammenzuarbeiten, das den BSI-Zertifizierungsprozess schon für andere DiGA bereits erfolgreich abgeschlossen hat. So können Sie sich durch praxisnahe Insights oft wochenlange Arbeit ersparen. Die Kosten hierfür sind verhältnismäßig überschaubar, sollten aber eingeplant werden.
Wir bei QuickBird Medical unterstützen Unternehmen in allen technischen und organisatorischen Aspekten der BSI-Zertifizierung. Wir haben u. a. mit der von uns entwickelten DiGA „Oriko®“ eines der ersten 3 BSI-Zertifikate überhaupt erhalten. Wir helfen Ihnen, effizient und sicher durch die Zertifizierung zu kommen. Kontaktieren Sie uns hierzu gern: Kontakt aufnehmen
4.2 Zeitrahmen für den Zertifizierungsprozesses
Der Zertifizierungsprozess und somit auch der Zeitplan können in die oben genannten drei Phasen gegliedert werden:
- Vorbereitungsphase: Hier implementiert Ihr Entwicklerteam die BSI-Vorgaben. Wie schnell Sie hierbei sind, kommt auf viele Faktoren an (Kompetenz der Entwickler, Zustand des aktuellen Systems in Bezug auf Security, Anzahl der Entwickler etc.)
- Prüfphase: Der Zeitrahmen hängt hier stark von der Prüfstelle und der Qualität Ihrer Dokumentation und Produktimplementierung ab. Sie sollten mit mindestens 4 bis 6 Wochen rechnen, bis Sie einen Prüfbericht erhalten.
- Zertifizierungsphase: Sobald die Prüfstelle den Prüfbericht für Ihr Produkt an das BSI geschickt hat, beginnt die Zertifizierungsphase. Auf offizieller Seite des BSI wird hier von 3 Wochen geredet. Unserer Erfahrung nach sollte man hier aktuell aber eher 6 Wochen ansetzen.
5. Gültigkeitsdauer & Rezertifizierung nach BSI TR-03161
Ein vom BSI erteiltes Zertifikat nach Technischen Richtlinien ist zeitlich befristet gültig. In der Regel beträgt die Gültigkeitsdauer eines Produktzertifikats fünf Jahre.
Abweichende Laufzeiten sind nur möglich, wenn dies im jeweiligen Zertifizierungsprogramm ausdrücklich festgelegt ist. Die Gültigkeit ist zudem immer auf die konkret geprüfte Produktversion sowie auf die zugrunde liegende Version der Technischen Richtlinie beschränkt.
Kann die Konformität eines Produkts nach Ablauf der Gültigkeit oder nach wesentlichen, sicherheitsrelevanten Änderungen nicht ohne erneute Prüfung bestätigt werden, ist eine Rezertifizierung erforderlich. Dabei wird erneut geprüft, ob das Produkt die Anforderungen der TR erfüllt. Der Prüfumfang kann sich auf die geänderten Teile beschränken, sofern frühere Prüfergebnisse weiterhin verwendbar sind.
Laut offiziellem Zitat des BSI hier, sollte eine Rezertifizierung spätestens drei Monate nach Ablauf des bestehenden Zertifikats beantragt werden (ob das BfArM dem für DiGA auch so zustimmt, bleibt abzuwarten). Bei erfolgreichem Abschluss wird ein neues Zertifikat für die geänderte Produktversion ausgestellt.
6. BSI TR-03161-Zertifikat mit Auflagen
Ein Zertifikat nach BSI TR-03161 hat üblicherweise eine Gültigkeit von bis zu fünf Jahren. In Einzelfällen kann das BSI jedoch im Rahmen der Zertifizierungsentscheidung Nebenbestimmungen in Form von Auflagen festlegen, die vom Hersteller innerhalb eines definierten Zeitraums umzusetzen sind.
In der Praxis bedeutet dies, dass ein Zertifikat bis zur Bearbeitung von bestimmten Auflagen mit einer Gültigkeit von wenigen Monaten erteilt werden kann. Der Hersteller erhält in diesem Fall zunächst einen gültigen Zertifizierungsnachweis (mit Auflagen), ist jedoch verpflichtet, die festgelegten Auflagen fristgerecht umzusetzen und nachzuweisen. Werden diese Auflagen nicht erfüllt, kann die Zertifizierung wieder aufgehoben werden. Bei nachweislicher Erfüllung der Auflagen innerhalb der Frist wird dann ein reguläres Zertifikat ohne Auflagen ausgestellt.
Dieses Vorgehen ermöglicht es, vereinzelte Abweichungen später nachzuziehen, um z. B. den DiGA-Antragsprozess nicht zu blockieren (bzw. vom BfArM abgelehnt zu werden). Ein BSI-Zertifikat mit Auflagen setzt aber eine umfassende Implementierung aller Anforderungen der BSI TR-03161 durch den DiGA-Hersteller voraus. Die Zertifizierung ist weiterhin enorm herausfordernd und sollte auf keinen Fall unterschätzt werden.
7. Änderungen an zertifizierten Produkten – BSI TR-03185
Ein Zertifikat gilt ausschließlich für die im Rahmen der Konformitätsprüfung geprüfte Produktversion. Jede Änderung am Produkt, etwa durch Weiterentwicklung, Updates, Patches oder Hotfixes, führt zunächst zu einer neuen Version, für die das bestehende Zertifikat keine automatische Gültigkeit mehr besitzt. Ob und wie diese Änderungen zertifizierungsseitig behandelt werden, hängt von ihrer Auswirkung auf die TR-Konformität ab.
Können Auswirkungen auf die Konformität nicht ausgeschlossen werden, ist eine Re-Zertifizierung erforderlich. Handelt es sich hingegen um geringfügige Änderungen, bei denen eine Beeinträchtigung der Konformität eindeutig ausgeschlossen werden kann, kommt ein sogenanntes Maintenanceverfahren infrage. In diesem Fall wird das bestehende Zertifikat auf die neue Produktversion erweitert, ohne dass eine erneute vollständige Konformitätsprüfung notwendig ist. Die Entscheidung darüber trifft das BSI auf Basis einer vom Hersteller vorzulegenden Änderungsdokumentation und Auswirkungsanalyse. Die ursprüngliche Laufzeit des Zertifikats bleibt dabei unverändert.
Zukünftiger Umgang mit Produktänderungen – die neue BSI TR-03185
Jede kleinere Änderung durch das BSI absegnen zu lassen, macht den DiGA-Entwicklungsprozess natürlich deutlich ineffizienter. Gerade auch bei kritischen Fehlern einer DiGA ist es wichtig, Probleme durch Hotfixes schnell zu beheben. Der aktuelle Prozess mit BSI-Prüfungen jeder Änderung scheint hierfür nicht geeignet.
Aktuell arbeitet das BSI daher an einer grundlegenden Weiterentwicklung des Umgangs mit Produktänderungen bei DiGAs. Im Mittelpunkt steht dabei die geplante Einführung der BSI TR-03185 „Sicherer Software-Lebenszyklus“, die den Update- und Änderungsprozess von Softwareprodukten strukturiert adressieren soll.
Mit der TR-03185 ist ein eigenständiges Zertifizierungsprogramm vorgesehen, bei dem Konformitätsprüfungen durch vom BSI akkreditierte IT-Grundschutz-Auditoren durchgeführt werden.
Die Idee dahinter: DiGA-Hersteller mit einer zusätzlichen Zertifizierung nach BSI TR-03185 haben nachweislich einen sicheren Software-Entwicklungsprozess (bzw. Lebenszyklus). Daher soll es diesen Herstellern erlaubt sein, bestimmte Änderungen am Produkt auch ohne vorherige BSI-Prüfung zu veröffentlichen.
Es ist vorgesehen, dass DiGA-Hersteller mit einer Zertifizierung nach BSI TR-03185 deutlich weniger Prüfverfahren für Folgeversionen ihrer nach BSI TR-03161 zertifizierten Produkte durchführen müssen. DiGA-Hersteller sind aber nicht explizit zur Zertifizierung nach BSI TR-03185 verpflichtet. Implizit wird der reibungslose Betrieb einer DiGA ohne die TR-03185-Zertifizierung aber vermutlich schwer möglich sein.
Die Inhalte der BSI TR-03185 können Sie hier einsehen: Link (BSI-Website)
Aktuell sind noch keine Antragstellungen oder Zertifizierungen möglich. Nach aktuellem Stand werden erste Zertifizierungen frühestens ab Anfang 2026 erwartet. Die konkreten Rahmenbedingungen hierzu befinden sich jedoch noch in Abstimmung.
8. Praktische Implikationen für DiGA
Die Anforderungen der BSI TR-03161 wirken sich unmittelbar auf das Produkt sowie die Entwicklung, den Betrieb und die Weiterentwicklung von DiGAs aus. Wir bei QuickBird Medical haben die BSI-Anforderungen mittlerweile in mehreren DiGA vollständig implementiert und z. B. für die DiGA „Oriko®“ eins der ersten 3 BSI-Zertifikate erhalten.
Die BSI-Zertifizierung hatte auf unsere Kundenprojekte viele Auswirkungen. Einige fassen wir hier zusammen:
- Änderungen nach der Zertifizierung: Anpassungen am Produkt sind später deutlich aufwendiger, da ein komplexer Änderungsprozess durchlaufen werden muss und Genehmigungen pro Release zusätzliche Zeit und Kosten verursachen.
- Stark regulierte Zwei-Faktor-Authentifizierung: Es existieren sehr hohe Anforderungen an die Umsetzung von Zwei-Faktor-Authentifizierung in DiGA. Diese Anforderungen beeinträchtigen je nach Implementierung stark die Nutzerfreundlichkeit der DiGA. Es gibt hier bessere und schlechtere Varianten der Umsetzung im Sinne der Nutzerfreundlichkeit. (Kontaktieren Sie uns gern, falls Sie hierzu Input benötigen.)
- Kurze Session-Laufzeiten und potenziell häufige Re-Logins: Die Anforderungen der BSI TR-03161 führen dazu, dass Anmelde- und Session-Zeiten stark begrenzt werden müssen. Je nach technischer Umsetzung kann dies dazu führen, dass Nutzer sich vergleichsweise häufig erneut anmelden müssen, was den Nutzungskomfort der DiGA spürbar beeinträchtigen kann. Auch hier gibt es Möglichkeiten, das Ganze so zu lösen, dass es den Nutzer weniger beeinträchtigt. (Kontaktieren Sie uns gern, falls Sie hierzu Input benötigen.)
- Erhöhter Entwicklungsaufwand: Architektur, Implementierung, Tests und Dokumentation erfordern deutlich mehr Aufwand als bei nicht regulierten Anwendungen.
- Datenverlust bei Geräteverlust: Es gibt verschiedene Wege, die TR-Anforderungen in einer DiGA umzusetzen. Jede davon hat Vor- und Nachteile. Bei bestimmten Varianten in Bezug auf Authentifizierung ist es nahezu unmöglich, die DiGA-Daten wiederherzustellen, wenn ein Nutzer z. B. sein Handy verliert.
- Strenge Resilience-Anforderungen: Bestimmte Betriebssystemversionen und Gerätekonfigurationen sind ausgeschlossen, etwa ältere Android-Versionen oder aktivierter Developer Mode, obwohl diese bei einem relevanten Anteil der Nutzer noch verbreitet sind. Das ist auf der einen Seite aus Sicherheitssicht nachvollziehbar, aber führt dazu, dass z. B. bis zu 10% Ihrer Zielgruppe keinen Zugriff auf die DiGA erhalten können.
- Eingeschränkte Hotfix-Fähigkeit: Kurzfristige Fehlerbehebungen sind offiziell nur mit Freigabe durch das BSI möglich, was schnelle Reaktionen erschwert. Dies soll sich perspektivisch durch die zusätzliche Zertifizierung nach TR-03185 verbessern, wodurch bestimmte Änderungen ggf. keine Freigabe mehr erfordern würden.
9. Verpflichtender Penetrationstest für DiGA
Die Pflicht zu einem Penetrationstest („Pen-Test“) ergibt sich nicht aus der BSI TR-03161. Sie stammt aus dem gesetzlichen Rahmen der DiGA-Verordnung (DiGAV). Dort wird festgelegt, dass jede DiGA einen aktuellen Penetrationstest nachweisen muss, um die Anforderungen an die Datensicherheit gemäß Anlage 1 DiGAV zu erfüllen.
Mit dem neuen DiGA-Leitfaden vom 10.12.2025 bezieht das BfArM hierzu konkrete Stellung: Durch die Zertifizierung nach TR-03161 entfällt die Notwendigkeit eines zusätzlichen Pen-Tests für die Zulassung einer DiGA.
10. BSI TR-03161 vs. ISO 27001
Für die Listung einer DiGA im Verzeichnis im BfArM muss der Hersteller sowohl eine ISO 27001-Zertifizierung als auch eine BSI TR-03161-Zertifizierung vorlegen. Bei beiden geht es um Informationssicherheit. Wieso also zwei Zertifizierungen?
Der Grund hierfür liegt im Fokus dieser beiden Normen.
Die ISO 27001 ist eine reine Managementnorm: Sie definiert, wie ein umfassendes Informationssicherheits-Managementsystem (ISMS) im Unternehmen aufgebaut, betrieben und kontinuierlich verbessert wird. Zertifiziert wird also das Unternehmen und seine Prozesse, nicht ein einzelnes Produkt. Es findet keine produktbezogene Prüfung, kein Architektur-Review und keine Analyse von Quellcode oder konkreten Sicherheitsmaßnahmen eines Produkts statt.
Die BSI TR-03161 hingegen ist eine technische Produktzertifizierung speziell für digitale Gesundheits- und Pflegeanwendungen. Sie legt sehr konkrete technische und organisatorische Sicherheitsanforderungen fest und prüft, ob das einzelne Produkt (inklusive App, Backend, Schnittstellen) diese erfüllt. Damit ist sie wesentlich spezifischer, strenger und produktnäher als ISO 27001 und dient in Deutschland als gesetzliche Grundlage für DiGA– und DiPA-Zulassungen.
| Kategorie | ISO 27001 | BSI TR-03161 |
| Art der Norm | Managementnorm | Technische Produktsicherheitsrichtlinie |
| Zertifizierungsobjekt | Unternehmen bzw. ISMS | Konkretes Produkt (App, Backend, Webportal) und speziell konkrete Produktversion |
| Fokus | Prozesse, Governance, Risikomanagement | Technische Sicherheitsanforderungen, Architektur, Implementierung |
| Reichweite | Firmenweit, unabhängig von Produkten | Produktbezogen und verpflichtend für DiGA/DiPA |
| Technische Tests | Keine spezifischen Produktprüfungen | Detaillierte technische Tests inkl. Kryptografie, APIs, Hosting |
| Quellcode-Analyse | Nicht Bestandteil | Fester Bestandteil der Prüfung |
| Anforderungen | Abstrakt, prozessorientiert | Konkrete, detaillierte Vorgaben für Produkt und Prozesse |
| Zielsetzung | Etablierung eines ISMS | Sicherer Betrieb und sichere Entwicklung einer Gesundheitsanwendung |
11. Kritik am Prozess
Sowohl die Verpflichtung zur Zertifizierung nach BSI TR-03161 als auch die daraus resultierenden, sehr strikten Anforderungen werden von vielen Seiten stark kritisiert. Häufig genannte Punkte sind dabei:
- Einschränkungen bei der Benutzerfreundlichkeit: Mehrstufige Sicherheitsmechanismen und strikte Vorgaben können die Usability spürbar beeinträchtigen. Es wird betont, dass Patienten dadurch einen erschwerten Zugang zu digitalen Gesundheitsangeboten haben.
- Eingeschränkte Zugänglichkeit für relevante Zielgruppen: Durch die sehr restriktiven Geräte- und Systemanforderungen der BSI TR-03161 werden viele Endgeräte ausgeschlossen. Insbesondere ältere Menschen sowie Personen mit körperlichen oder kognitiven Einschränkungen nutzen häufig Geräte, die diese Anforderungen nicht erfüllen und somit nicht akzeptiert werden. Dadurch bleibt ein erheblicher Teil der eigentlich relevanten Zielgruppe von der Nutzung digitaler Gesundheitsanwendungen ausgeschlossen.
- Hoher Aufwand in Umsetzung und Dokumentation: Die sehr detaillierten Sicherheits- und Prozessanforderungen führen nach Ansicht zahlreicher Hersteller zu erheblichen zusätzlichen Entwicklungs- und Dokumentationsarbeiten. Monatelange Zusatzarbeiten erschweren und verhindern derartige neue Innovationen.
- Steigende Kosten für Anbieter: Sowohl die Zertifizierung selbst als auch der kontinuierliche Nachweis der Konformität verursachen enorme Kosten aufseiten der DiGA-Hersteller. Gerade für kleine Unternehmen sind diese Kosten schwer bis unmöglich zu finanzieren.
- Unklarheit bei den Anforderungen: Bei vielen Punkten der TR-03161 ist nicht eindeutig, wie eine korrekte Umsetzung nach Ansicht des BSI auszusehen hat. Teilweise widersprechen sich einzelne Anforderungen auch gegenseitig. Somit stecken DiGA-Hersteller viel Zeit in eine Implementierung, die dann potenziell nach langen Wartezeiten durch das BSI abgewiesen wird.
- Nationaler Alleingang: Kritiker bemängeln, dass die BSI TR-03161 einen rein deutschen Sicherheitsstandard darstellt, der nicht mit europäischen Vorgaben harmonisiert ist. Dies erschwert die internationale Skalierung und erhöht den regulatorischen Aufwand für Hersteller.
Mehrere Branchenvertretungen haben diese Punkte in ihren Stellungnahmen hervorgehoben und eine ausgewogenere Lösung zwischen Sicherheit, Praktikabilität und wirtschaftlicher Umsetzbarkeit gefordert (siehe z. B. hier, hier und hier).
Inwiefern auf diese Punkte in Zukunft eingegangen wird, bleibt abzuwarten. Hier ist vor allem das nächste Update zur BSI TR-03161 relevant, welches vermutlich im Jahr 2026 erscheinen soll.
12. Wichtige Links und Ressourcen
12.1 Allgemeine Informationen
Hier finden Sie wichtige Ressourcen in Bezug auf die BSI-Zertifizierung:
- Links zu den PDFs der BSI TR-03161 (siehe unterer Teil der Seite): Link (BSI-Website)
- FAQ zur BSI TR-03161: Link (BSI-Website)
- Informationen zu TR-Zertifizierungen beim BSI: Link (BSI-Website)
- Links zu den PDFs der BSI TR-03161 (Sicherer Software-Lebenszyklus): Link (BSI-Website)
12.2 Liste der bereits nach TR-03161 zertifizierten Anwendungen
Sie können in unserem DiGA-Verzeichnis-Analyzer bequem nach den BSI-zertifizierten DiGA filtern, die jeweilige Prüfstelle der DiGA sehen und weitere Attribute vergleichen: DiGA-Verzeichnis-Analyzer für Hersteller (inkl. BSI-Zertifizierungen)
Die offizielle Liste der digitalen Gesundheitsanwendungen (inkludiert Anwendungen vor Listung), die bereits ein Zertifikat vom BSI erhalten haben, finden Sie hier: Offizielle Liste der zertifizierten Anwendungen
12.3 Liste der Prüfstellen für die BSI TR-03161
Hier finden Sie eine Liste der vom BSI akkreditierten Prüfstellen: Liste der akkreditierten TR-Prüfstellen
Wenn Sie eine Textsuche auf der Seite nach „03161“ durchführen, finden Sie alle Stellen, welche Ihre Zertifizierung nach BSI TR-03161 begleiten können.
13. Zukunftsausblick für die BSI TR-03161 und DiGA-Datensicherheitszertifizierung
Die Anforderungen an Datensicherheit für DiGAs entwickeln sich aktuell weiter und werden in den kommenden Jahren weiter konkretisiert. Dabei zeichnen sich insbesondere drei relevante Entwicklungen ab, die von DiGA-Herstellern frühzeitig eingeplant werden sollten.
- Aktualisierung der BSI TR-03161: Für das kommende Jahr ist eine Aktualisierung der TR-03161 angekündigt. Erwartet wird, dass dabei praktische Erfahrungen aus der bisherigen Anwendung einfließen und einzelne Anforderungen präzisiert oder angepasst werden.
- Einführung der BSI TR-03185 zum sicheren Software-Lebenszyklus: Ergänzend zur TR-03161 ist mit der TR-03185 eine neue Zertifizierung für den gesamten Software-Lebenszyklus in Vorbereitung. Ziel ist es, Update- und Änderungsprozesse systematischer abzubilden und perspektivisch den Aufwand für wiederkehrende Re-Zertifizierungen bei Folgeversionen zu reduzieren.
- Datenschutz-Zertifizierung: Neben Datensicherheit ist weiterhin eine Datenschutz-Zertifizierung für DiGA geplant (und bereits gesetzlich verankert). Sobald ein Prüfprozess und Prüfstellen hierfür etabliert wurden, wird das BfArM diese Zertifizierung von DiGA-Herstellern einfordern.
14. Fazit
Für DiGAs steigt die Zahl verpflichtender Zertifizierungen scheinbar kontinuierlich weiter: Medizinprodukt-Zulassung bzw. Zertifizierung, ISO 27001, BSI TR-03161 und zukünftige Zertifizierungen wie die BSI TR-03185 (nicht verpflichtend, aber scheinbar unumgänglich) und das Datenschutz-Zertifikat für DiGA.
Dies führt zu einer zunehmenden regulatorischen Komplexität, die insbesondere kleinere Hersteller stark belastet. Das notwendige Budget zur Vorbereitung, Umsetzung und kontinuierlichen Erhaltung dieser Zertifizierungen wächst aktuell stetig weiter. Aus unserer Sicht besteht die Gefahr, dass Innovationsgeschwindigkeit und Markteintritt neuer Lösungen dadurch gebremst oder sogar verhindert werden.
Dennoch ist das Ganze mit der richtigen Strategie (und ausreichendem Budget) weiterhin machbar. Wir haben in unseren DiGA-Projekten gelernt, mit der Komplexität umzugehen, und viele Learnings gemacht, die wir gerne weitergeben. Gerade in Bezug auf die BSI TR-03161-Zertifizierung fehlen bei einigen Anforderungen klare Vorgaben zur Umsetzung von offizieller Seite. Damit nicht jeder Hersteller diese Learnings neu machen muss, beraten wir Hersteller bei der Umsetzung der BSI-Vorgaben.
Falls Sie einen Partner benötigen, der Ihre DiGA implementiert, die BSI-Anforderungen umsetzt und alle weiteren regulatorischen Pflichten für Sie übernimmt: Kontaktieren Sie uns gerne. Wir waren bereits in die Umsetzung von über 15 verschiedenen DiGA-Projekten involviert und besitzen die notwendigen Zertifikate, um Ihre DiGA sicher auf den Markt zu bringen. Mehr Infos dazu finden Sie hier.
The post BSI TR-03161 für DiGA: Zertifizierung der Datensicherheit appeared first on QuickBird Medical.
]]>The post DiGA Leitfaden: Historie aller Änderungen appeared first on QuickBird Medical.
]]>Der aktuelle DiGA-Leitfaden des BfArM ist hier abrufbar.
Inhaltsverzeichnis
- 1. Änderungen zwischen der Version 3.6 & Version 3.5 des DiGA-Leitfadens
- 2. Änderungen zwischen der Version 3.5 & Version 3.4 des DiGA-Leitfadens
- 3. Zukünftige Änderungen des DiGA-Leitfadens
1. Änderungen zwischen der Version 3.6 & Version 3.5 des DiGA-Leitfadens
Die Aktualisierung des DiGA-Leitfadens vom 10.12.2025 (von Version 3.5 auf 3.6) ist umfangreich. Der Leitfaden ist von 177 auf 215 Seiten angewachsen und viele relevante Themenblöcke sind hinzugekommen.
Die größeren Änderungen inkludieren z. B.:
- DigiG: Neue Regelungen des DigiG wurden nun auch im DiGA-Leitfaden eingearbeitet (z. B. für DiGA der Risikoklasse IIb)
- DiGAV Update: Neue Regelungen des Referentenentwurfs der 2. DiGAV-ÄndV wurden eingearbeitet (vorwiegend anwendungsbegleitende Erfolgsmessung – AbEM)
- BSI TR-03161 als Standard: Datensicherheit wird vollständig über das BSI-Zertifikat abgedeckt. Der Nachweis der Datensicherheit über die DiGAV-Checkliste entfällt
- DiGA-Qualifizierung präzisiert: Klarere Abgrenzung zwischen DiGA und Nicht-DiGA inkl. neuem Entscheidungsbaum zur DiGA-Fähigkeit (Zweckbestimmung, Individualisierung, Steuerung aktiver Medizinprodukte, Risikoklasse IIb)
- Evidenzanforderungen konkretisiert: Präzisierungen zu Studiendesign, Signifikanzniveau (5 %), Randomisierung, Studienberichten und Auswertung
- Interoperabilität aktualisiert: Ablösung von „vesta“ durch „INA“ als Referenz für interoperable E-Health-Infrastruktur
- Prozessklarstellungen: Detailliertere Regelungen zu Fristen, Antragsportal, Lebenszyklus, Pflichten nach Listung und Löschung
- Umfang & Struktur erweitert: Allgemein deutlicher Ausbau des Leitfadens (177 → 215 Seiten) mit neuen Kapiteln, FAQs und Beispielen
Dazu kommen viele weitere kleinere bis mittelgroße Änderungen. Wir haben alle Änderungen analysiert und für Sie untenstehend zusammengefasst. Aufgrund des Umfangs der Änderungen geben wir hier nicht den genauen Wortlaut wieder, sondern fassen die Anpassungen jedes geänderten Kapitels grob zusammen.
Für interne Produktentscheidungen sollten Sie allerdings den Originalwortlaut im DiGA-Leitfaden nachlesen. Die folgende Liste soll Ihnen vor allem eine strukturierte Übersicht geben.
1.1 PDF-Dokument mit allen Änderungen (von Version 3.5 zu 3.6)
Zur einfacheren Durchsicht aller Änderungen können Sie auch das folgende Dokument nutzen. Dieses Dokument ist das Ergebnis eines automatisierten Vergleichs der beiden Versionen (3.6 vs. 3.5) des DiGA-Leitfadens.
Download der Gegenüberstellung aller Änderungen
1.2 Liste aller Änderungen (von Version 3.5 zu 3.6)
Abkürzungsverzeichnis
Folgende Abkürzungen wurden hinzugefügt:
- AbEM: Anwendungsbegleitende Erfolgsmessung
- CGI Scale: Clinical Global Impressions Scale
- DigiG: Digital-Gesetz
- J2R: Jump-To-Reference
- PGI-C: Patient Global Impression of Change
- PREMs: Patient-Reported Experience Measures
- PROMs: Patient-Reported Outcome Measures
- RCT: Randomisierte kontrollierte Studie
- SOP: Standard Operating Procedure
- INA: Interoperabilitäts-Navigator der Gematik
2.1 Was ist eine DiGA und was nicht?:
Präzisere Abgrenzung zwischen DiGA und Nicht-DiGA (Individualisierung, Konsistenz der Zweckbestimmung, Steuerung aktiver therapeutischer Medizinprodukte etc.); neuer „DiGA-Fähigkeit Entscheidungsbaum“ als Grafik; Inklusion von Risikoklasse IIb nach DigiG
2.1.1.1 Erstattungsfähigkeit von Hardware (neues Kapitel):
Klarstellung, dass Gebrauchsgegenstände des täglichen Lebens (z. B. Smartwatches, Smartphones, Tablets) sowie Hilfsmittel nach § 33 SGB V nicht über DiGA erstattungsfähig sind; Präzisierung der Gerätekategorien (keine, enthaltene, optionale, notwendige Zusatzgeräte)
2.1.1.2 Wahlfreiheit der Versicherten und ärztliche Therapiefreiheit (neues Kapitel):
Klarstellung, dass DiGA keine Lock-in-Effekte erzeugen dürfen und grundsätzlich mit vergleichbaren Hilfsmitteln nutzbar sein müssen; Ausnahme bei zwingender Bindung an ein spezifisches, nicht gelistetes Medizinprodukt aus Sicherheitsgründen.
2.1.2 Kombination mit Dienstleistungen:
Konkretes neues Beispiel, welches sich nicht als DiGA qualifizieren würde (DiGA für reine Datenübermittlung an ärztliches Personal)
2.1.3 Umfang der DiGA:
Konkretisierung, dass Nachweis für positiven Versorgungseffekt ohne optionale Funktionen erbracht werden muss und optionale Funktionen gekennzeichnet werden müssen
2.3 Vorläufige und dauerhafte Aufnahme:
Klarstellung, dass DiGA der MDR-Risikoklasse IIb nicht vorläufig bzw. auf Erprobung gelistet werden können.
2.3.1 Beantragung der dauerhaften Aufnahme in das DiGA-Verzeichnis:
Ergänzungen für DiGA der MDR-Risikoklasse IIb
3.1 Aufbau der Checklisten für die Anforderungen an DiGA:
Datensicherheit wird nicht mehr nach DiGAV-Checkliste nachgewiesen, sondern durch BSI-Zertifikat.
3.2 Sicherheit und Funktionstauglichkeit:
Konkretisierung, dass Konformitätsbewertungsverfahren für das Medizinprodukt vor DiGA-Antragsstellung abgeschlossen sein muss
3.3 Datenschutz:
Datenschutz-Zertifizierung aktuell weiterhin noch nicht möglich; Verweis auf zukünftige Pflicht der Zertifizierung, sobald dies möglich ist
3.3.2 Zulässige Datenverarbeitung nach § 4 Absatz 2 Satz 1 und 2 DiGAV:
Ergänzung von FAQ-Element zu Einwilligung bei externen Datenplattformen (z.B. Apple Health)
3.3.4 Ausblick auf die Datenschutzkriterien nach § 139e Absatz 11 SGB V:
Aktualisierung des aktuellen Stands in Bezug auf zukünftige Datenschutz-Zertifizierung (aktuell weiterhin noch nicht möglich)
3.4 Datensicherheit:
Viele Inhalte entfernt und ersetzt durch Referenzen zu BSI TR-03161 und dem damit einhergehenden Zertifizierungsprozess; DiGAV-Checkliste zur Datensicherheit muss nicht mehr ausgefüllt werden (stattdessen Referenz auf TR-03161 etc.)
3.4.2 Sicherheit als Prozess:
Klarstellung, dass das BSI-Zertifikat die Datensicherheit vollständig abdeckt und Pen-Test für den DiGA-Antrag nicht zusätzlich benötigt wird.
3.5.1.1 INA und MIOs als Basis einer interoperablen E-HealthInfrastruktur:
„vesta“ wird durch „INA“ abgelöst (mehr Infos hier)
3.5.4.3 Datenerfassung über Medizingeräte, Wearables und andere Sensorik:
„vesta“ wird durch „INA“ abgelöst (mehr Infos hier)
3.5.4.4 Schreiben der DiGA in die ePA/Implementierung der GesundheitsID:
Konkretisierungen für DiGA im Antragsverfahren (oder davor)
4.1.2 Patientenrelevante Struktur- und Verfahrensverbesserungen (pSVV):
Umfangreiche Klarstellungen, Beispiele und FAQ in Bezug auf pSVV
4.3 Allgemeine Anforderungen an Studien zum Nachweis positiver Versorgungseffekte:
Ergänzung des Mindest-Signifikanzniveaus von 5%; Ergänzungen zum Umgang mit Abbrechern bzw. Drop-outs, Sprachversion, Daten aus dem Ausland sowie Interventionszeitraum und Verordnungszeitraum
4.3.2 Grundsätze bei der Auswertung und Bewertung von Studienergebnissen durch den Hersteller (neues Kapitel):
Klarstellung, dass die Bewertung auf präspezifizierten, konservativen Analysen (insbesondere RCT-Zwischengruppenvergleiche) beruhen muss; Post-hoc-Analysen nur explorativ zu werten, vollständige Berücksichtigung der Kontrollgruppe sowie Einbezug von Konsistenz und klinischer Relevanz.
4.3.3 Auswahl und Begründung des Versorgungspfades (neues Kapitel):
Klarstellung, dass der Versorgungspfad das Studiendesign bestimmt, inkl. klarer Abgrenzung von alleiniger Therapie, Überbrückung und Add-on-Szenarien, vollständiger Dokumentation der Begleittherapien und ggf. Subgruppenanalysen bei heterogener Standardversorgung.
4.3.4 Anforderungen an Randomisierungsverfahren (neues Kapitel):
Konkretisierung der Anforderungen an eine valide, nicht manipulierbare Randomisierung inkl. Präspezifikation im Protokoll, dokumentierter Stratifizierung, geeigneter IT-Systeme, auditierbarer Verfahren, Zugriffsbeschränkungen und Maßnahmen zur Vermeidung von Vorhersehbarkeit (z. B. variable Blocklängen).
4.3.5 Studiendesign und methodische Planung (neues Kapitel):
Konkretisierung robuster Studiendesigns (u. a. RCT-Anforderungen), konservative Imputation fehlender Werte (z. B. J2R), präzise ITT-Definition sowie Pflicht zur vollständigen Dokumentation und Berichterstattung aller Abweichungen vom Studienprotokoll
4.3.6 Antragsunterlagen (neues Kapitel):
Klarstellung der einzureichenden Versionen (vor/nach Studienstart), verpflichtende Änderungsnachweise (Track Changes & Übersichtstabelle) sowie formale Anforderungen an Vollständigkeit und Dateiformat.
4.3.7 Inhaltliche und qualitative Anforderungen an Studienberichte (neues Kapitel):
Konkretisierung der Ergebnisdarstellung (Originalskalen, KI, p-Werte, Effektschätzer); verpflichtende Einordnung der klinischen Relevanz; detaillierte Drop-out-Analyse; klare Trennung der Analysepopulationen; hohe Anforderungen an Transparenz, Tabellen, Grafiken und Konsistenz der Berichterstattung
4.6.2 DiGA mit diagnostischer Komponente:
Konkretisierungen, was eine DiGA mit diagnostischer Komponente sein darf; neue FAQs
4.5.1 Begründung der Versorgungsverbesserung und systematische Datenauswertung:
Kleinere Ergänzungen zur Studien-Fallzahlplanung und statistischer Signifikanz
5 Ablauf des Verfahrens:
Ergänzung, dass Bescheide über Antragsportal übermittelt werden
5.1 Fristen für Antragsteller und BfArM:
Konkretisierungen zu Mängelschreiben und Fristen
5.2 Lebenszyklus einer DiGA im Verzeichnis:
Listung im Portal 5 Tage nach positivem Bescheid
5.2.2 Pflichten des Herstellers nach Aufnahme einer DiGA in das DiGA-Verzeichnis:
Pflicht zur vorzeitigen Anzeige von PZN-Änderungen
5.2.3 Verordnung einer DiGA:
Pflicht zur vorzeitigen Anzeige von PZN-Änderungen
5.2.5 Löschen einer DiGA aus dem DiGA-Verzeichnis:
Ergänzung der Mitteilungspflicht an Nutzer und Datenlöschung
5.4 Beratung durch das BfArM:
Empfehlung für frühzeitige Beratung bei Risikoklasse IIb-DiGA und DiGA mit Telemonitoring
6. Anwendungsbegleitende Erfolgsmessung (AbEM) (neues Kapitel):
Umfangreiches neues Kapitel zur AbEM aus dem Referentenentwurf der 2. DiGAV-ÄndV; detaillierte Definition der Datenpunkte, Berichtszeiträume und Fristen; Erklärung der geplanten Ausbaustufen der AbEM

Weitere kleinere Änderungen
Minimale Änderungen sind außerdem in den folgenden Kapiteln passiert: 2.3.4, 3.5.4, 4.3.8, 4.4.2, 5.2.4, 5.5
Diese sind größtenteils redaktionelle Optimierungen oder minimale Ergänzungen. Wir haben diese daher in der obigen Auflistung der Übersichtlichkeit halber weggelassen.
Abonnieren Sie unseren Newsletter, um über zukünftige Änderungen des DiGA-Leitfadens stets informiert zu bleiben.
2. Änderungen zwischen der Version 3.5 & Version 3.4 des DiGA-Leitfadens
Legende: Farbliche Veranschaulichung der Änderungen
In den Screenshots, die der jeweiligen Version des DiGA-Leitfadens entnommen sind, sind alle Änderungen farblich hinterlegt. Für die Markierung der Änderungsart werden die folgenden Farben verwendet.
| Farbe | Art der Änderung |
| Ergänzungen | |
|
Streichungen |
Änderungen im Kapitel 2.2.1.3 „Informationen für Leistungserbringende“
Der vorletzte Punkt in der Auflistung „Zur Verordnung relevante Informationen“ wurde um den Zusatz „und festgelegter Höchstbetrag“ ergänzt.
Neue Version 3.5:

Alte Version 3.4:
reine Ergänzung neuer Informationen – keine Entsprechung in alter Version
Änderungen im Kapitel 3.4.2 „Sicherheit als Prozess“
Im FAQ-Bereich wurde die Anforderung, Penetrationstests als Teil des Sicherheitsprozesses durchzuführen, verschärft.
Neue Version 3.5:

Alte Version 3.4:
reine Ergänzung neuer Informationen – keine Entsprechung in alter Version
Ergänzung eines neuen Kapitels 3.5.4.4 „Schreiben der DiGA in die ePA/Implementierung der GesundheitsID“:
Die neue Anforderung für alle Hersteller, nämlich das Schreiben der DiGA in die ePA sowie die Implementierung der GesundheitsID, wird erläutert. Die Umsetzungsfrist hierfür endet am 30.04.2024.
Neue Version 3.5:

Alte Version 3.4:
keine Entsprechung in alter Version, da neues Kapitel
Änderungen im Kapitel 4.2.2 „Angabe des positiven Versorgungseffektes“:
Mit einem neuen Hinweis wird die Anforderung des BfArM bezüglich des Nachweises mehrerer positiver Versorgungseffekte präzisiert. Demnach ist es für DiGA-Hersteller erforderlich, zunächst mindestens einen positiven Versorgungseffekt nachzuweisen, zu dem sie im Rahmen der vorläufigen Aufnahme durch einen BfArM-Bescheid verpflichtet wurden, bevor weitere positive Versorgungseffekte berücksichtigt werden können.
Neue Version 3.5:

Alte Version 3.4:
reine Ergänzung neuer Informationen – keine Entsprechung in alter Version
Änderungen im Kapitel 4.3 „Verwendung von DiGA-Daten“:
Die Möglichkeit, eine Analyse eigener, bereits verordneter DiGA während des Erprobungszeitraums für die Einreichung von Nachweisen zur endgültigen Aufnahme zu verwenden, wurde konkretisiert. Es ist gestattet, eine Analyse eigener, nah an der Versorgung durchgeführter Daten aus dem Erprobungszeitraum als Unterstützung für die Nachweisführung zur dauerhaften Aufnahme zu nutzen. Dabei ist wichtig sicherzustellen, dass das Vorgehen im Voraus festgelegt wurde und ausführlich in einem separaten Studienbericht beschrieben ist.
Neue Version 3.5:

Alte Version 3.4:

Änderungen im Kapitel 4.3.3 „Übertragbarkeit von Studienergebnissen“:
Im Abschnitt „Durchführung der Studie in Deutschland“ hat das BfArM drei Kriterien festgelegt, um die Übertragbarkeit der Studienergebnisse auf den Versorgungskontext in Deutschland zu belegen.
Neue Version 3.5:

Alte Version 3.4

Änderungen im Kapitel 5.2.2 „Änderung der DiGA-Bezeichnung“:
Hier wird darauf hingewiesen, dass eine Änderung der Bezeichnung einer DiGA eine wesentliche Veränderung darstellt und beim BfArM anzuzeigen ist.
Neue Version 3.5:

Alte Version 3.4:
reine Ergänzung neuer Informationen – keine Entsprechung in alter Version
Änderungen im Kapitel 5.2.3 „Kurzbezeichnung und PZN“:
Ein neuer Hinweis im Unterbereich „Pharmazentralnummer“ wurde hinzugefügt: Bei der Änderung der Kurzbezeichnung einer DiGA wird eine neue Pharmazentralnummer (PZN) vergeben.
Neue Version 3.5:

Alte Version 3.4:
reine Ergänzung neuer Informationen – keine Entsprechung in alter Version
Änderungen im Kapitel 5.2.4 „Verweis auf Übergangsfristen“
Die neuen Fristen für den Übergang sind im Abschnitt 3.5.4.4 verzeichnet.
Neue Version 3.5:

Alte Version 3.4:
reine Ergänzung neuer Informationen – keine Entsprechung in alter Version
3. Zukünftige Änderungen des DiGA-Leitfadens
Wir bringen diesen Artikel mit jeder Aktualisierung des DiGA-Leitfadens auf den neuesten Stand.
Abonnieren Sie unseren Newsletter, um über zukünftige Änderungen des DiGA-Leitfadens stets informiert zu bleiben.
The post DiGA Leitfaden: Historie aller Änderungen appeared first on QuickBird Medical.
]]>The post IVDR Software: Qualifizierung und Klassifizierung appeared first on QuickBird Medical.
]]>Diese Verordnung regelt Produkte, die Proben außerhalb des Körpers untersuchen, zum Beispiel Blut-, Urin- oder Gewebeproben. Der Begriff „in vitro“ bedeutet wörtlich „im Glas“ und beschreibt genau diese Untersuchungen außerhalb des menschlichen Körpers.
Dieser Artikel richtet sich an (angehende) Hersteller von IVD-Produkten, welche entweder komplett oder teilweise aus Software bestehen. Wir erklären Ihnen, wann Software unter die In Vitro Diagnostic Regulation (IVDR) fällt, wie die Qualifizierung funktioniert und nach welchen Kriterien die Risikoklassifizierung erfolgt.
Aus unserer praktischen Erfahrung in Kundenprojekten zur Umsetzung von IVDR-Software (und sogar mobilen IVD-Apps) können wir sagen: Die Einordnung ist nicht immer ganz einfach. Wir brechen das Thema für Sie daher herunter und teilen es in praxisnahe Unteraspekte auf. So erhalten Sie eine klare Grundlage, um die Qualifizierung und Klassifizierung Ihres Produkts vorzunehmen.
Inhaltsverzeichnis
- 1. Qualifizierung: Fällt Ihre Software unter die IVDR?
- 2. Klassifizierung: Unter welche Risikoklasse fällt Ihre Software?
- 3. Wer trifft die finale Entscheidung zur Qualifizierung & Klassifizierung?
- 4. Gemeinsamkeiten und Unterschiede: MDR vs. IVDR
- 5. Fazit
1. Qualifizierung: Fällt Ihre Software unter die IVDR?
Die Qualifizierung dient dazu, herauszufinden, ob Ihre Software überhaupt als In-vitro-Diagnostikum gilt und damit unter die IVDR fällt. Die Basis hierfür stellt die Zweckbestimmung (mehr dazu hier) Ihres Produkts dar.
Für die Qualifizierung sollten Sie folgende 3 Aspekte für Ihr Produkt prüfen.
1.1 Aspekt 1: Definition eines IVD nach IVDR
Zusammengefasst fällt Ihre Software in den regulierten Bereich der IVDR, wenn diese in die Definition eines „In-vitro-Diagnostikum“ nach IVDR Artikel 2(2) fällt. Der Wortlaut dieser Definition ist der Folgende:
„In-vitro-Diagnostikum“ bezeichnet ein Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibrator, Kontrollmaterial, Kit, Instrument, Apparat, Gerät, Software oder System — einzeln oder in Verbindung miteinander — vom Hersteller zur In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben, einschließlich Blut- und Gewebespenden, bestimmt ist und ausschließlich oder hauptsächlich dazu dient, Informationen zu einem oder mehreren der folgenden Punkte zu liefern
a) über physiologische oder pathologische Prozesse oder Zustände,
b) über kongenitale körperliche oder geistige Beeinträchtigungen,
c) über die Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit,
d) zur Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern,
e) über die voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf oder
f) zur Festlegung oder Überwachung therapeutischer Maßnahmen.
Prüfen Sie daher genau, ob Sie die Zweckbestimmung Ihres Produkts in dieser Definition wiederfinden.
1.2 Aspekt 2: Definition eines Medizinprodukts nach MDR
Im ersten Satz der obigen Definition sieht man, dass ein IVD auch ein „Medizinprodukt“ sein muss:
„In-vitro-Diagnostikum“ bezeichnet ein Medizinprodukt, das als Reagenz […]
Die IVDR verweist für die Definition eines Medizinprodukts auf die Medical Device Regulation (MDR). Daher müssen Sie zusätzlich zur obigen Definition auch die Definition eines Medizinprodukts mit der Zweckbestimmung Ihres Produkts abgleichen.
Wie genau die Definition eines Medizinprodukts nach MDR aussieht und wie Sie entscheiden, ob Sie mit Ihrem Produkt in diese Definition fallen, erfahren Sie in unserem Leitfaden: Ist Ihre Software ein Medizinprodukt?
Wichtig: Obwohl Sie unter die Definition eines Medizinprodukts nach MDR fallen, müssen Sie für ein In-Vitro-Diagnostikum nur die IVDR einhalten. Es gilt nicht gleichzeitig die MDR und die IVDR für Ihr Produkt.
1.3 Aspekt 3: Embedded, Zubehör oder Standalone-Software?
Neben der Beschäftigung mit der IVD-Definition, ist es aber vor allem auch wichtig, zu verstehen, wie die „regulatorische Architektur“ Ihres Gesamtprodukts aussieht:
- Wird die Software als Teil Ihres Hardware-Geräts mit-zertifiziert?
- Ist die Software gesondert zu behandeln und wird demnach als eigenes IVD-Produkt zertifiziert?
- Ist die Software ein Zubehör zu dem eigentlichen IVD-Produkt (z. B. Hardware)?
Die Antworten auf diese Fragen beeinflussen stark den Zulassungsprozess Ihres Produkts und sollten daher bereits zu Beginn der Entwicklung gefunden werden.
Hierfür müssen Sie entscheiden, in welche der folgenden drei Kategorien Ihre Software-Komponente fällt:
| Kategorie | Erklärung | Auswirkungen |
| #1: Teil eines IVD-Geräts | Software ist fest mit einem IVD-Gerät verbunden und steuert dieses oder beeinflusst seine Nutzung. Sie übernimmt Funktionen, die für den Betrieb des Geräts notwendig sind. | Software und Gerät werden als ein zusammengehöriges Medizinprodukt zertifiziert. |
| #2: Zubehör für ein IVD-Gerät | Software ist selbst kein IVD, wird aber genutzt, um die Funktion eines IVD zu unterstützen. Sie ermöglicht oder verbessert die Nutzung eines anderen IVD. Siehe Definition von „Zubehör“ in der IVDR. | Zubehör wird unabhängig vom eigentlichen IVD, mit dem es verwendet wird, gesondert klassifiziert. |
| #3: Standalone-IVD-Software | Software arbeitet unabhängig von einem physischen Gerät und erzeugt unabhängig diagnostische Informationen, basierend auf Proben- oder Messdaten. | Software wird als getrenntes IVD nach IVDR zertifiziert. |
1.4 Zusammenfassung: Aspekte der Qualifizierung
Für Sie gilt es also herauszufinden:
- Ist Ihre Software Teil eines Geräts, Zubehör oder Standalone-Software?
- Fällt Ihre Software unter die Definition eines Medizinprodukts nach MDR?
- Fällt Ihre Software unter die Definition eines In-Vitro-Diagnostikums nach IVDR?
Sie sollten diese Themen zu Beginn Ihres Vorhabens durchlaufen, um die Anwendbarkeit der IVDR für Ihr Produkt zu prüfen.
1.5 Schritt-für-Schritt-Anleitung zur Qualifizierung
Die Antwort auf die oben genannten Kernfragen ist nicht immer ganz eindeutig. Daher gibt es zusätzliche Guidance-Dokumente zur IVDR (und MDR), die einen offiziellen Charakter haben.
Bezüglich der Qualifizierung von Software in Bezug auf die IVDR sollten Sie konkret einen Blick auf die „MDCG 2019-11“ werfen. Diese versucht u.a., den Entscheidungsprozess im Rahmen der IVDR von Software für Sie zu vereinfachen bzw. zu konkretisieren: Link zur MDCG Guidance
Guidance-Dokumente der MDCG sind zwar genau genommen nicht rechtlich bindend, werden aber in der Praxis auch meist von benannten Stellen herangezogen, um derartige Entscheidungen im Rahmen der Zertifizierung zu treffen.
Wir haben den Entscheidungsbaum der MDCG 2019-11 auf Deutsch übersetzt und die Formulierungen zur besseren Verständlichkeit etwas vereinfacht:
Entscheidungsbaum zur Qualifizierung: Fällt Ihre Software unter die IVDR?
Schritt 1: Ist Ihre Software ein Medizinprodukt nach MDR?
Fallen Sie mit der Zweckbestimmung Ihrer Software in die Definition eines Medizinprodukts nach MDR? Hierfür lohnt sich ein Blick auf unseren Leitfaden zu genau diesem Thema: Leitfaden: Ist Ihre Software ein Medizinprodukt?
Schritt 2: Stellt Ihre Software Informationen bereit, die in den Anwendungsbereich der Definition eines In-vitro-Diagnostikums fallen?
Konkret formuliert: Stellt Ihre Software Informationen zu einem der folgenden Zwecke zur Verfügung? (siehe Definition eines IVD in der IVDR):
a) über physiologische oder pathologische Prozesse oder Zustände,
b) über kongenitale körperliche oder geistige Beeinträchtigungen,
c) über die Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit,
d) zur Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern,
e) über die voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf oder
f) zur Festlegung oder Überwachung therapeutischer Maßnahmen.
Schritt 3: Erzeugt Ihre Software Informationen ausschließlich auf Grundlage von Daten, die von In-vitro-Diagnostika gewonnen wurden?
Sind Sie sicher, dass die Daten nicht auch aus anderen Quellen wie z. B. einem Medizinprodukt (kein IVD) stammen? Nur dann können Sie diese Frage mit „Ja“ beantworten.
Schritt 4: Wird die Zweckbestimmung im Wesentlichen durch Datenquellen bestimmt, die von In-vitro-Diagnostika stammen?
Wenn es auch Informationen gibt, die nicht von einem IVD-Produkt stammen: Wird die Zweckbestimmung dann trotzdem im Wesentlichen durch IVD-Informationen bestimmt? Wie stark beeinflussen die „Nicht-IVD-Informationen“ denn die Zweckbestimmung?
Ergebnis: Sind Sie im Entscheidungspfad unten beim „Ergebnis IVDR“ angelangt?
Dann ist Ihre Software höchstwahrscheinlich ein IVD oder Teil eines IVDs, das unter die IVDR fällt. Leider hat natürlich jede Frage und jede Definition Platz für verschiedene Auslegungen. Insofern sollten Sie diese Entscheidung nicht voreilig treffen und sich im Zweifel professionelle Beratung zu diesem Thema einholen.
Hinweis: Unsere Grafik dient nur der Veranschaulichung. Für die finale Qualifizierung des Produkts empfehlen wir Ihnen, mit der englischen Version der MDCG-Guidance im Original-Wortlaut zu arbeiten. Sie können uns auch gerne kontaktieren, falls Sie Input für die Qualifizierung oder Klassifizierung Ihres Produkts benötigen. Wir entwickeln IVDR- und MDR-Software auf Auftragsbasis und geben gerne praktische Insights weiter.
1.6 Qualifizierung: Beispiele für IVDR und Nicht-IVDR
Um das Thema etwas greifbarer zu machen, finden Sie hier einige Beispiele für Software-Produkte, die laut MDCG-Guidances tendenziell …
- … ein IVD nach IVDR sind.
- … kein IVD sind, aber ein Medizinprodukt nach MDR sind.
- … weder IVD noch Medizinprodukt sind.
Qualifizierungsbeispiel: IVD nach IVDR
Ein deutliches Beispiel für ein IVD ist Software, die Rohdaten aus Labortests automatisch auswertet. Zum Beispiel:
- Software, welche die optische Dichte eines ELISA-Lesegeräts analysiert. Bei einem ELISA entsteht durch eine chemische Reaktion in der Probe ein Farbwechsel. Das Gerät schickt Licht durch die Probe und misst, wie viel Licht absorbiert wird. Aus dieser Farbintensität kann abgeleitet werden, wie viel von einem bestimmten Stoff im Blut vorhanden ist.
- Software, die Muster eines sogenannten Blots interpretieren. Ein Blot ist ein Labortest, bei dem Proteine auf eine Membran übertragen werden und dort sichtbare Linien oder Punkte bilden. Diese Muster zeigen an, ob bestimmte Antikörper oder Eiweiße vorhanden sind.
Solche Software nimmt die Messdaten auf und berechnet daraus mithilfe von medizinischen Algorithmen einen diagnostischen oder prognostischen Befund. Deshalb gilt sie in der Regel als IVD-Software.
Ein weiteres spannendes Beispiel, das wir bei QuickBird Medical gut kennen, ist die App „Accu-Chek SugarView“ von Roche. Die Accu-Chek SugarView App bestimmt den Blutzuckerbereich, indem sie den Accu-Chek Active Teststreifen und zwei Fotos, die mit der Smartphone-Kamera vom Teststreifen aufgenommen werden, auswertet. Die intelligente Software funktioniert ohne jegliches zusätzliches Glukosemessgerät. Sie wurde als IVD-Software klassifiziert und hat die CE-Kennzeichnung erhalten (mehr dazu finden Sie hier)
Qualifizierungsbeispiel: Kein IVD, sondern Medizinprodukt nach MDR
Es gibt auch Software, die zwar einen medizinischen Zweck erfüllt, aber keine Laborproben auswertet. Solche Anwendungen fallen nicht unter die IVDR, sondern unter die MDR. Dazu gehören insbesondere Programme, die:
- medizinische Bilder verarbeiten oder analysieren, zum Beispiel in der Radiologie oder Dermatologie
- Messwerte eines medizinischen Geräts bewerten, solange diese Werte nicht aus In-Vitro-Tests stammen
Wenn die Software keine Blut-, Urin- oder Gewebeproben untersucht, aber trotzdem medizinische Entscheidungen unterstützt, handelt es sich dann eben womöglich um ein Medizinprodukt nach MDR und nicht um ein IVD.
Qualifizierungsbeispiel: Kein IVD und kein MDR-Medizinprodukt
Viele Softwareprodukte erfüllen weder die Definition eines IVD nach IVDR noch die eines Medizinprodukts nach MDR. Darunter fallen zum Beispiel:
- Laborinformationssysteme (LIS), die nur Daten verwalten oder Laborprozesse organisieren
- Programme, die Ergebnisse einfach darstellen, zum Beispiel in Form von Diagrammen oder einfachen Statistiken
- Software, die Messdaten lediglich speichert oder weiterleitet, ohne diese zu interpretieren oder zu analysieren
Solche Software trifft keine eigenen diagnostischen Aussagen und hat keinen direkten medizinischen Zweck. Deshalb fällt sie weder unter die IVDR noch unter die MDR.
2. Klassifizierung: Unter welche Risikoklasse fällt Ihre Software?
„Ja, ich will!“ – Können Sie diese Aussage mit Selbstbewusstsein in Bezug auf die IVD-Qualifizierung (siehe oben) machen? Fallen Sie also in den Anwendungsbereich der IVDR? (für viele Hersteller vermutlich kein „Ich will“, sondern eher ein „Ja, ich muss 😔“). Dann wird es nun Zeit, sich mit den Risikoklassen der IVDR zu beschäftigen.
Um die regulatorischen Anforderungen für Sie als Hersteller und Ihr Produkt zu bestimmen, müssen Sie erstmal wissen, in welche Risikoklasse Ihr Produkt bzw. die Komponenten Ihres Produkts fallen.
2.1 Welche Risikoklassen gibt es unter IVDR?
Die IVDR unterscheidet zwischen den folgenden Risikoklassen: A, B, C, D
Die Idee ist, dass man Produkten mit höherem Risiko (z.B. B, C oder D) auch strengere Anforderungen auferlegt als Produkten mit sehr geringem Risiko (Klasse A).
Hinweis: Damit unterscheidet sich die Risikoklassifizierung unter IVDR von Medizinprodukten, die unter die Medical Device Regulation (MDR) fallen. Die MDR unterteilt Medizinprodukte in die Risikoklassen I, IIa, IIb und III. Mehr dazu in unserem MDR-Leitfaden.
Recherche in der EUDAMED-Datenbank: Anzahl der IVDR-Produkte & IVDR-Software-Produkte pro Risikoklasse
(Beschreibung der Risiken jeweils nach IMDRF-Guidance)
In der Abbildung finden Sie das Ergebnis unserer Recherchen zur Risikoklassen-Verteilung von IVDR-Produkten innerhalb der EUDAMED-Datenbank. Vorsicht: Die Statistik hat keinerlei Aussagekraft in Bezug auf Ihr Produkt. Die Tatsache, dass die meisten Produkte in Klasse A eingeordnet sind, hat viele Gründe. Sie müssen trotzdem strukturiert evaluieren, in welche Risikoklasse Ihre Software fällt (und tatsächlich ist es gerade bei diagnostischer Software vermutlich nicht die Klasse A oder B).
2.2 Die wichtigsten Implikationen der Risikoklassen der IVDR
Die Risikoklasse eines IVD-Software-Produkts wirkt sich in vielerlei Hinsicht auf Entwicklung, Zulassungsprozess und Post-Market-Aktivitäten aus. Hier fassen wir die zentralen Aspekte für Sie zusammen.
Deckungsgleiche Anforderungen an das Qualitätsmanagement & technische Dokumentation
Je nach Risikoklasse, werden unterschiedlich strenge Anforderungen im Rahmen der IVDR gestellt. Dies betrifft aber tatsächlich nicht die sogenannten Grundlegenden Sicherheits- und Leistungsanforderungen an das IVD-Produkt. Die grundlegenden Anforderungen an das Qualitätsmanagementsystem des Herstellers und die technische Dokumentation des Produkts sind bei allen Risikoklassen (A, B, C und D) identisch (bei den höheren Risikoklassen C & D greifen aber vereinzelt Zusatzanforderungen).
Hierbei sind besonders relevant:
- Qualitätsmanagement nach ISO 13485 → Unser Leitfaden zur ISO 13485, speziell für Software-Produkte
- Software-Lebenszyklus nach IEC 62304 → Unser Leitfaden zur IEC 62304
- Risikomanagement nach ISO 14971
- Gebrauchstauglichkeit nach IEC 62366-1
Benannte Stelle ab Risikoklasse B
Der wohl größte Unterschied ist, dass Hersteller von Produkten der Klasse B, C oder D (im Gegensatz zu den meisten Produkten der Klasse A) durch einen Zertifizierungsprozess mit einer benannten Stelle gehen müssen, bevor die Produkte auf den Markt gelangen können. Eine akkreditierte Stelle muss also u.a. die technische Dokumentation und das Qualitätsmanagementsystem prüfen und freigeben.
Das ist übrigens sehr ähnlich zur Regelung unter Medical Device Regulation (MDR), wo Klasse I-Produkte quasi vom Hersteller selbst zertifiziert werden und ohne externe Prüfung auf den Markt gehen können (im Gegensatz zu Klasse IIa, IIb und III unter MDR).
Unterschiede zwischen Klasse B, C und D
Der Unterschied zwischen Risikoklasse A und B ist somit klar (die Prüfung der benannten Stelle). Gibt es ab Risikoklasse B dann jedoch keine weiteren Anforderungen mit steigender Risikoklasse (C und D)? Wofür dann 4 Klassen und nicht nur eine?
Antwort: Doch, die gibt es. Vereinfacht gesprochen, bei Risikoklasse C und D:
- Es werden vereinzelt weitere Dokumentationsanforderungen gestellt (z. B. ein zusätzlicher Kurzbericht über Sicherheit und Leistung).
- Der Detailgrad der Prüfung durch die benannte Stelle steigt mit steigender Risikoklasse.
- Bei Risikoklasse D wird ein Referenzlabor der Europäischen Union in die Prüfungen und Zulassung einbezogen.
Eine gute Übersicht bekommen Sie auch, wenn Sie im offiziellen Gesetzestext nach dem Stichwort „Klasse“ suchen und einmal alle Erwähnungen überprüfen.
2.3 Einordnung in die richtige Klasse: Regeln der IVDR
Wichtig ist zuallererst, dass die gesamte Klassifizierung auf Basis der Zweckbestimmung Ihres Produkts stattfinden muss. Wie genau Sie die Zweckbestimmung für ein Medizinprodukt definieren, erklären wir in unserem gesonderten Leitfaden hier.
Die Klassifizierung von IVDR-Produkten wird durch eine überraschend übersichtliche, kurze Liste von 7 Regeln bestimmt (ANHANG VIII der IVDR). Statt diese hier redundant in einigen Worten wiederzugeben, empfehlen wir Ihnen stark, sich einfach selbst einmal den kurzen Abschnitt in der IVDR hierzu durchzulesen (siehe unten). Wir haben den genauen Wortlaut aus der Verordnung für Sie im nächsten Kapitel eingefügt. Das ist tatsächlich auch der einzige Abschnitt der IVDR, der sich mit der Einordnung in die Risikoklasse beschäftigt.
2.3.1 Auszug der EU-Verordnung: Klassifizierungsregeln nach Anhang VIII der IVDR
Die folgenden Klassifizierungsregeln finden Sie in der Verordnung über In-Vitro-Diagnostika (IVDR) in Anhang VIII:
2. KLASSIFIZIERUNGSREGELN
2.1. Regel 1
Produkte mit den folgenden Zweckbestimmungen werden der Klasse D zugeordnet:
— Nachweis des Vorhandenseins von oder der Exposition gegenüber übertragbaren Erregern in Blut, Blutbestandteilen, Zellen, Geweben oder Organen oder in einem ihrer Derivate, um ihre Eignung für die Transfusion, Transplantation oder Zellgabe zu bewerten;
— Nachweis des Vorhandenseins von oder der Exposition gegenüber übertragbaren Erregern, die eine lebensbedrohende Krankheit mit einem hohen oder mutmaßlich hohen Verbreitungsrisiko verursachen;
— Bestimmung der Infektionslast einer lebensbedrohenden Krankheit, dessen Überwachung im Rahmen des Patientenmanagements von entscheidender Bedeutung ist.2.2. Regel 2
Produkte, die zur Blutgruppenbestimmung oder zur Feststellung einer feto-maternalen Blutgruppen-Inkompatibilität oder zur Gewebetypisierung verwendet werden, um die Immunkompatibilität von für die Transfusion, Transplantation oder Zellgabe bestimmtem Blut, Blutbestandteilen, Zellen, Geweben oder Organen festzustellen, werden der Klasse C zugeordnet, es sei denn, sie werden zur Bestimmung eines der folgenden Marker eingesetzt:
— ABNull-System [A (ABO1), B (ABO2), AB (ABO3)];
— Rhesus-System [RH1 (D), RHW1, RH2 (C), RH3 (E), RH4 (c), RH5 (e)];
— Kell-System [Kel1 (K)];
— Kidd-System [JK1 (Jka), JK2 (Jkb)];
— Duffy-System [FY1 (Fya), FY2 (Fyb)].
In diesem Fall werden sie der Klasse D zugeordnet.2.3. Regel 3
Produkte werden der Klasse C zugeordnet, wenn sie folgende Zweckbestimmung haben:
a) Nachweis des Vorhandenseins von oder Exposition gegenüber einem sexuell übertragbaren Erreger;
b) Nachweis eines Infektionserregers ohne hohes oder mutmaßlich hohes Verbreitungsrisiko im Liquor oder im Blut;
c) Nachweis eines Infektionserregers, wenn ein signifikantes Risiko besteht, dass ein fehlerhaftes Ergebnis den Tod oder eine ernste gesundheitliche Schädigung der getesteten Person, des getesteten Fötus oder Embryos oder der Nachkommen der getesteten Person verursacht;
d) Feststellung des Immunstatus von Frauen gegenüber übertragbaren Erregern zum Zwecke des pränatalen Screenings;
e) Feststellung des Vorliegens einer Infektionskrankheit oder des Immunstatus, wenn das Risiko besteht, dass ein fehlerhaftes Ergebnis zu einer Patientenmanagemententscheidung führen würde, die eine lebensbedrohende Situation für den Patienten oder die Nachkommen des Patienten schafft;
f) Einsatz als therapiebegleitende Diagnostika;
g) Einsatz zur Stadieneinteilung einer Krankheit, wenn das Risiko besteht, dass ein fehlerhaftes Ergebnis zu einer Patientenmanagemententscheidung führen würde, die eine lebensbedrohende Situation für den Patienten oder die Nachkommen des Patienten schafft;
h) Einsatz zur Krebsvorsorge, -diagnose oder -stadieneinteilung;
i) Durchführung von Gentests beim Menschen;
j) Überwachung des Niveaus von Arzneimitteln, Stoffen oder biologischen Komponenten, wenn das Risiko besteht, dass ein fehlerhaftes Ergebnis zu einer Patientenmanagemententscheidung führen würde, die eine lebensbedrohende Situation für den Patienten oder die Nachkommen des Patienten schafft;
k) Management von Patienten, die an einer lebensbedrohenden Krankheit leiden oder deren Zustand lebensbedrohend ist;
l) Untersuchung auf genetisch bedingte Störungen beim Embryo oder Fötus;
m) Untersuchung auf genetisch bedingte Störungen bei Neugeborenen, wenn der fehlende Nachweis und die fehlende Behandlung dieser Störungen zu lebensbedrohenden Situationen oder ernsten gesundheitlichen Schädigungen führen können.2.4. Regel 4
a) Produkte zur Eigenanwendung werden der Klasse C zugeordnet, ausgenommen Produkte zur Feststellung einer Schwangerschaft, zur Fertilitätsuntersuchung und zur Bestimmung des Cholesterinspiegels und Produkte zum Nachweis von Glukose, Erythrozyten, Leukozyten und Bakterien im Urin, die der Klasse B zugeordnet werden.
b) Produkte zur Verwendung in patientennahen Tests werden für sich allein klassifiziert.2.5. Regel 5
Die folgenden Produkte werden der Klasse A zugeordnet:
a) Erzeugnisse für den allgemeinen Laborbedarf, Zubehör ohne kritische Merkmale, Pufferlösungen, Waschlösungen sowie allgemeine Nährmedien und histologische Färbungen, die vom Hersteller dafür vorgesehen sind, die Produkte für In-vitro-Diagnoseverfahren im Zusammenhang mit einer spezifischen Untersuchung einsetzbar zu machen;
b) Instrumente, die vom Hersteller speziell für die Verwendung bei In-vitro-Diagnoseverfahren vorgesehen sind;
c) Probenbehältnisse.2.6. Regel 6
Produkte, die nicht unter die zuvor beschriebenen Klassifizierungsregeln fallen, werden der Klasse B zugeordnet.2.7. Regel 7
Produkte, bei denen es sich um Kontrollen ohne einen zugewiesenen quantitativen oder qualitativen Wert handelt, werden der Klasse B zugeordnet.
2.3.2 Auszug der EU-Verordnung: Durchführungsvorschriften für die Klassifizierung
Neben den Klassifizierungsregeln finden Sie in der IVDR noch einige Durchführungsvorschriften, welche die Klassifizierung betreffen. Untenstehend finden Sie den Auszug aus der IVDR im Anhang VIII.
1. DURCHFÜHRUNGSVORSCHRIFTEN
1.1. Die Anwendung der Klassifizierungsregeln richtet sich nach der Zweckbestimmung der Produkte.
1.2. Wenn das betreffende Produkt dazu bestimmt ist, in Verbindung mit einem anderen Produkt angewandt zu werden, werden die Klassifizierungsregeln auf jedes Produkt gesondert angewendet.
1.3. Zubehör für ein In-vitro-Diagnostikum wird unabhängig von dem Produkt, mit dem es verwendet wird, gesondert klassifiziert.
1.4. Software, die ein Produkt steuert oder dessen Anwendung beeinflusst, wird derselben Klasse zugerechnet wie das Produkt. Ist die Software von anderen Produkten unabhängig, so wird sie für sich allein klassifiziert.
1.5. Kalibratoren zur Verwendung mit einem Produkt werden derselben Klasse zugerechnet wie das Produkt.
1.6. Kontrollmaterialien mit quantitativen oder qualitativen zugeordneten Werten, die für einen spezifischen oder mehrere Analyten bestimmt sind, werden derselben Klasse zugerechnet wie das Produkt.
1.7. Der Hersteller berücksichtigt alle Klassifizierungs- und Durchführungsregeln, um die angemessene Einstufung des Produkts festzustellen.
1.8. Wenn der Hersteller für ein Produkt mehrere Zweckbestimmungen angibt und das Produkt daher mehr als einer Klasse zuzuordnen ist, wird es in die höchste der Klassen eingestuft.
1.9. Für den Fall, dass für dasselbe Produkt mehrere Klassifizierungsregeln gelten, wird die Regel der Einstufung in die höchste dieser Klassen angewendet.
1.10. Jede Klassifizierungsregel gilt für erstmalige Tests, Bestätigungstests und Ergänzungstests.
2.4 MDCG-Guidance zur Klassifizierung nach IVDR
Nicht jeder Satz der oben gezeigten Klassifizierungsregeln der IVDR ist selbsterklärend und eindeutig. Daher gibt es zusätzliche Leitfäden der MDCG, die Ihnen u.a. bei der Bestimmung der Risikoklasse helfen sollen.
Für die Klassifizierung von Software-Produkten gibt es zwei relevante MDCG-Guidance-Dokumente:
- Allgemeine Guidance – „MDCG 2020-16: Guidance on Classification Rules for in vitro Diagnostic Medical Devices under
Regulation (EU) 2017/746“: Dieses Dokument liefert zu jeder Klassifizierungsregel der IVDR hilfreiche Beispiele und Anmerkungen. So erhalten Sie zusätzliche Unterstützung bei der Evaluation, ob bestimmte Klassifizierungsregeln auf Ihr Produkt anwendbar sind. –> Link - Software-spezifisch für MDR und IVDR – „MDCG 2019-11: Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 IVDR“: Das Dokument geht u.a. auf die Besonderheiten der Qualifizierung und Klassifizierung von Software ein. Im Gegensatz zur MDCG 2020-16 ist der Umfang der relevanten Abschnitte verhältnismäßig knapp. –> Link
Hinweis: Die Formulierung „Regulation (EU) 2017/746“ steht für die IVDR. „Regulation (EU) 2017/745“ steht hingegen für die MDR.
2.5 Klassifizierung: Software-Beispiele für alle Klassen der IVDR
Auch für die Klassifizierung möchten wir Ihnen einige Software-Beispiele aus der MDCG-Guidance an die Hand geben, um die Thematik zu verdeutlichen. Natürlich sind dies nur Veranschaulichungen und der Teufel steckt manchmal im Detail. Insofern sollten Sie die Klassifizierung Ihres Produkts nicht rein auf dem Vergleich mit existierenden Beispielen vornehmen, sondern alle Regeln der IVDR einzeln prüfen.
Software-Beispiel für Risikoklasse A
Eine Software gehört vor allem zur Risikoklasse A, wenn sie ausschließlich dazu gedacht ist, ein IVD-Gerät (nach IVDR) zu steuern, welches ebenfalls in Klasse A eingestuft ist. Das ergibt sich aus Anhang VIII 1.4 der IVDR: „Software, die ein Produkt steuert oder dessen Anwendung beeinflusst, wird derselben Klasse zugerechnet wie das Produkt.“
Ein konkretes Beispiel ist eine Software, die einen CRP-Analyser (C-reaktives-Protein-Gerät), der unter die Klasse A fällt, aus der Ferne bedient. Die Software trifft keine eigenen diagnostischen Aussagen, sondern sorgt nur dafür, dass das Gerät funktioniert. Deshalb wird sie in dieselbe Klasse wie das Gerät eingestuft, hier also Klasse A.
Software-Beispiel für Risikoklasse B
Etwas schwierig ist es, offizielle Beispiele für Software der Klasse B zu finden. (Software-)Produkte landen nur in Klasse B, falls sie in spezielle Produktkategorien zur Eigenanwendung (z.B. Schwangerschaftstests) fallen (Regel 4) oder sonst keine der anderen Klassifizierungsregeln greift (Regel 6).
In der Guidance „MDCG 2020-16¡ findet sich folgendes Beispiel:
„Blood gas analyser and the software driving and influencing it are classified as […] Class B (Rule 6) where the blood gas analyser directly measures a parameter from a specimen (in the absence of a reagent or disposable sensor cassette) AND where an erroneous result in a measured parameter WILL NOT LEAD TO a patient management decision resulting in a lifethreatening situation.“
Software-Beispiel für Risikoklasse C
Software, die in einem automatisierten ELISA-System HbA1c-Werte berechnet und zur Diabetes-Diagnose oder Verlaufskontrolle verwendet wird, fällt unter die Klasse C. Das lässt sich aus Regel 3(k) der IVDR ableiten.
Software, die in einem automatisierten PAP-Screening-System Abstriche als auffällig oder unauffällig klassifiziert, fällt auch unter Klasse C. Das lässt sich aus Regel 3(h) der IVDR ableiten.
Software-Beispiel für Risikoklasse D
Software, die die Ergebnisse eines automatisierten Line-Immunoassays für HIV-Antikörper interpretiert, fällt unter die Klasse D. Das lässt sich aus Regel 1 der IVDR ableiten. In diesem Fall bewertet die Software hochkritische Infektionsmarker.
3. Wer trifft die finale Entscheidung zur Qualifizierung & Klassifizierung?
Sie als Hersteller des Produkts müssen diese Entscheidung treffen. Die Verantwortung dafür, die Anwendbarkeit der IVDR auf Ihr (Software-)Produkt zu prüfen (Qualifizierung) sowie die Einstufung in die passende Risikoklasse, liegt initial komplett bei Ihnen.
Dabei ist eine sorgfältige Bewertung entscheidend, denn eine benannte Stelle kann die Zertifizierung ablehnen, wenn diese zu einer anderen Einschätzung bei der Risikoklassifizierung kommt. Außerdem sind Sie natürlich rechtlich angreifbar, falls Sie diese Einstufung nicht richtig vornehmen.
Gerade bei einer Einstufung in die Risikoklasse A sollten Sie vorsichtig sein, da die Klassifizierung hier initial nicht von einer benannten Stelle geprüft wird. Somit haben Sie noch weniger gefühlte Sicherheit bei der Auswahl der Risikoklasse.
Außerdem: Obwohl hier keine benannte Stelle eingebunden wird, kann die zuständige Aufsichtsbehörde eine fehlerhafte Klassifizierung bemerken und im schlimmsten Fall verlangen, dass das Produkt vom Markt genommen wird. Auch Produkte der Risikoklasse A werden in unregelmäßigen Abständen Prüfungen unterzogen. Des Weiteren wird im Falle eines Personenschadens durch Ihr Produkt sicherlich auch rechtlich hinterfragt, ob die von Ihnen als Hersteller zugewiesene Risikoklasse angemessen war.
Wenn Sie unsicher sind, lohnt es sich, eine unabhängige Einschätzung einzuholen. Mögliche Anlaufstellen sind:
- ein spezialisierter Rechtsanwalt, der ein Gutachten erstellen kann
- ein Regulatory-Expert oder Berater mit entsprechender Erfahrung
- das BfArM (in der Praxis aufgrund monatelanger Bearbeitungszeiten allerdings schwierig)
QuickBird Medical entwickelt IVDR- und MDR-Software für Kunden auf Auftragsbasis. Gerne geben wir Ihnen auf Basis unserer Erfahrung Input zur Klassifizierungs- und Qualifizierungsentscheidung. Bei Bedarf können wir Ihnen z. B. auch einen Anwalt aus unserem Kontaktkreis vermitteln, der für Sie ein Gutachten erstellen kann.
→ Kontakt aufnehmen
Wichtig: Weder eine externe Einschätzung noch ein formelles Gutachten bieten eine vollständige rechtliche Absicherung. Sie können im Falle eines Rechtsstreits jedoch als wichtige Entscheidungsgrundlage dienen.
4. Gemeinsamkeiten und Unterschiede: MDR vs. IVDR
Die MDR regelt klassische Medizinprodukte, die meist direkt am Patienten eingesetzt werden, während die IVDR In-vitro-Diagnostika betrifft, also Produkte, die z. B. Proben wie Blut oder Gewebe untersuchen.
Beide Verordnungen funktionieren in vielen Bereichen ähnlich: Hersteller brauchen ein Qualitätsmanagementsystem, eine technische Dokumentation, eine verantwortliche Person für die Regelkonformität und müssen ihre Produkte nach festen Risikoklassen einstufen.
Größere Unterschiede liegen u.a. in den folgenden Punkten:
- Unterschiedliche Klassifizierungslogik: Die Risikoklassen und die Klassifizierungslogik der IVDR und MDR unterscheiden sich stark. Hier ist auch zu beachten, dass Software-Produkte unter IVDR deutlich seltener in die niedrigste Klasse (A) fallen, als dies bei MDR mit Klasse I der Fall ist. Somit ist bei den meisten IVD-Produkten mit Software eine benannte Stelle involviert.
- Spezifischere Leistungsbewertung bei IVDs: IVD-Produkte müssen eine sehr umfangreiche Performance Evaluation durchlaufen. Diese umfasst unter anderem die wissenschaftliche Validität, analytische Performance (z. B. Sensitivität, Spezifität) und die klinische Performance. Auch unter MDR gibt es Medizinprodukte, die derartige Validierungsanforderungen einhalten müssen (z. B. ein Produkt zur Diagnose von Krankheiten auf Basis von CT-Bilddaten). Diese Anforderungen sind in der IVDR jedoch deutlich konkreter definiert als in der MDR.
- Zusätzliche Prüfmechanismen bei Hochrisiko-IVDs: Produkte der IVDR-Klasse D (z. B. HIV- oder Hepatitis-Tests) müssen durch EU-Referenzlabore (EURLs) unabhängig überprüft werden.
5. Fazit
IVDR-Software bewegt sich im Spannungsfeld zwischen technischer Entwicklung, Laborrealität und strengen regulatorischen Anforderungen.
Dieser Artikel soll Ihnen helfen, die ersten zentralen Fragen zu beantworten: Fällt Ihre Software überhaupt unter die IVDR und in welche Risikoklasse gehört sie? Wenn diese Eckpfeiler stehen, haben Sie eine solide Basis, um Qualitätsmanagement, die weitere Dokumentation, Validierung und Zusammenarbeit mit einer benannten Stelle gezielt anzugehen.
Im Zuge der IVDR ist zu beachten, dass der Großteil aller Software-basierten Diagnose-Produkte nicht in Risikoklasse A fallen wird. Daher sollten Sie damit rechnen, dass Sie als Hersteller eine benannte in den Zulassungsprozess inkludieren müssen. Die Kosten und den zusätzlichen Zeitaufwand für diese Zertifizierung sollten Sie in Ihrer Produktplanung berücksichtigen.
Über uns: QuickBird Medical entwickelt IVDR- und MDR-Software für Kunden auf Auftragsbasis. Wir übernehmen sowohl die technische Umsetzung als auch die regulatorische Konformität Ihres (Software-)Produkts. Wenn Sie also ein IVDR- oder MDR-Produkt planen, welches Software-Komponenten enthält, kontaktieren Sie uns gern für ein unverbindliches Erstgespräch.
→ Kontakt aufnehmen
In Zukunft planen wir weitere Artikel, die Ihnen bei der Planung und Entwicklung von IVDR-Produkten bzw. speziell der Software-Komponente dieser helfen sollen. In unserem Newsletter informieren wir Sie über neue Artikel und Änderungen zu diesem Thema.
The post IVDR Software: Qualifizierung und Klassifizierung appeared first on QuickBird Medical.
]]>The post DiPA-Leitfaden: digitale Anwendungen für die Pflege appeared first on QuickBird Medical.
]]>Mit dem Digitale‑Versorgung‑und‑Pflege‑Modernisierungs‑Gesetz (DVPMG) wurde im Juni 2021 auch ein Pendant dazu für die Pflege geschaffen: digitale Pflegeanwendungen (DiPA).
DiPA sollen den Alltag pflegebedürftiger Menschen und deren Umfeld durch Web- und Smartphone-Apps unterstützen. Beispielsweise könnte eine digitale Pflegeanwendung dazu beitragen, das Sturzrisiko von Pflegebedürftigen zu minimieren, oder die Kommunikation zwischen Angehörigen und Pflegekräften zu verbessern.
Die Verordnung zur Prüfung der Erstattungsfähigkeit digitaler Pflegeanwendungen (DiPAV) gibt Aufschluss darüber, wie das DiPA-Antragsverfahren abläuft und welche Anforderungen an DiPA gestellt werden, um in das Verzeichnis für digitale Pflegeanwendungen aufgenommen zu werden.
In diesem Artikel erklären wir den Unterschied zwischen der DiPA und DiGA und behandeln die wichtigsten Inhalte der DiPAV und haben letztlich einen Leitfaden für das DiPA-Verfahren erstellt. Dieser zeigt auf, welchen Prozess Ihre Anwendung durchlaufen muss, um in das BfArM-Verzeichnis aufgenommen zu werden.
Inhaltsverzeichnis
- 1. Aktuellste Entwicklungen: DiPA im Gesetz zur Befugniserweiterung und Entbürokratisierung in der Pflege (BEEP)
- 2. Abgrenzung der DiPA zur DiGA
- 3. Leitfaden für die DiPAV
- 4. Weitere Wege in die Kostenerstattung durch Krankenkassen
- 5. Fazit
1. Aktuelle Entwicklungen: DiPA im Gesetz zur Befugniserweiterung und Entbürokratisierung in der Pflege (BEEP)
Wichtig: Im vorliegenden Artikel beschreiben wir die aktuell geltende gesetzliche Regelung zu DiPA. Allerdings wurde im November 2025 das BEEP-Gesetz (Gesetz zur Befugniserweiterung und Entbürokratisierung in der Pflege) vom Bundestag beschlossen, das in seiner jetzigen Fassung relevante Änderungen bezüglich DiPA vorsieht.
Das BEEP-Gesetz sollte zum ersten Januar 2026 in Kraft treten, wurde aber aufgrund kurzfristiger Ergänzungen zur Krankenhausfinanzierung vom Bundesrat in den Vermittlungsausschuss verwiesen. Sollte es unverändert in Kraft treten, wären wichtige Verbesserungen für DiPA enthalten:
- Ein Erprobungsjahr für DiPA soll analog zum DiGA-Fast-Track eingeführt werden. Voraussetzung ist ein entsprechendes Evaluationskonzept, da der vollständige Nutzennachweis noch nicht vorliegt. Gleichzeitig wird die Erforderlichkeitsprüfung für ergänzende Unterstützungsleistungen durch das BfArM gestrichen.
- Vergütungsverhandlungen mit dem GKV-Spitzenverband können parallel laufen. Der Anspruch auf Versorgung startet, sobald die Vergütung vereinbart ist. Diese gilt dann rückwirkend ab dem Tag der Verzeichnisaufnahme.
- Der Versorgungsanspruch pflegebedürftiger Personen wird von bisher 50 € pro Monat auf 70 € erhöht. Davon sollen 40 € auf die DiPA selbst entfallen, 30 € können gegebenenfalls für ergänzende Unterstützungsleistungen erstattet werden. Allerdings ist zu beachten, dass dieses monatliche Budget eventuell bereits bei Inanspruchnahme einer einzigen DiPA aufgebraucht ist. Da aber theoretisch mehrere DiPA parallel angewendet werden können, könnte es nach dieser Regelung weiterhin dazu kommen, dass nur die erste Anwendung erstattet wird und jede weitere DiPA von den Pflegenden oder Gepflegten aus eigener Tasche bezahlt werden muss.
- Fokus ist die Unterstützung von Pflegenden: Der Nachweis eines pflegerischen Nutzens für die pflegebedürftige Person durch DiPA-Hersteller ist nicht mehr zwingend notwendig. Wenn eine Entlastung der Pflegenden durch die DiPA erreicht wird, wird davon ausgegangen, dass sich dies positiv auf die Pflegebedürftigen auswirkt. Um diese Entlastung nachzuweisen, muss eine Evaluation vorgelegt werden. Wie diese aussehen kann, ist nicht näher definiert.
- DiPA sollen künftig nicht nur entlasten, sondern auch gezielt dazu beitragen, dass sich der Zustand pflegebedürftiger Menschen nicht weiter verschlechtert, also präventiv wirken und vorhandene Selbstständigkeit stabilisieren.
- Die Anwendung von DiPA bleibt auf den Bereich der ambulanten häuslichen Pflege beschränkt, der Einsatz in der stationären Pflege ist nicht vorgesehen.
Hinweis: Da das BEEP-Gesetz noch nicht in Kraft getreten ist und sich auch noch inhaltliche Änderungen ergeben können, aktualisieren wir den Artikel, sobald es neue Erkenntnisse gibt. Im Folgenden informieren wir über die aktuell geltende gesetzliche Regelung zu DiPA.
2. Abgrenzung der DiPA zur DiGA
Um die digitale Pflegeanwendung besser zu verstehen, hilft es, ihren Einsatz und Inhalt von der DiGA abzugrenzen.
DiGA sind Apps, die erkrankten Menschen bei der Therapie, Diagnostik, Kompensation oder Linderung von Krankheiten, Verletzungen und Behinderungen unterstützen. Ihr Einsatz wurde am 19. Dezember 2019 mit dem DVG gesetzlich festgelegt. Seitdem können DiGA von Krankenkassen als Teil der ärztlichen und psychotherapeutischen Behandlung erstattet werden. Die DiPA ist ein ähnliches Konzept, das sich aber in erster Linie an pflegebedürftige Menschen richtet. Sie fand Mitte 2021 durch das Digitale-Versorgung-und-Pflege-Modernisierungs-Gesetz (DVPMG) Einzug ins elfte Sozialgesetzbuch (SGB).

Unterschied zwischen DiGA und DiPA
Da die Abgrenzung zwischen DiGA und DiPA auf den ersten Blick nicht ganz offensichtlich ist, möchten wir in den folgenden Kapiteln die wichtigsten Unterschiede beleuchten.
2.1 Krankenkasse vs. soziale Pflegekasse
Den ersten großen Unterschied zwischen DiGA und DiPA finden wir schon über ihren Platz im Gesetzestext. DiGA werden im SGB V – Gesetzliche Krankenversicherung, DiPA im SGB XI – Soziale Pflegeversicherung definiert. Eine DiGA wird demnach von der Krankenkasse erstattet, wohingegen für die Kosten einer DiPA die soziale Pflegekasse aufkommt.
Eine DiGA wird von ÄrztInnen und PsychotherapeutInnen verschrieben und dient explizit dazu, Menschen mit bestimmten Indikationen bei Linderung, Therapie und Diagnostik zu unterstützen. Die Voraussetzung, um eine DiGA von der Krankenkasse erstattet zu bekommen, ist ähnlich wie bei Medikamenten die Verschreibung eines Arztes oder Psychotherapeuten. Auch eine direkte Genehmigung der Krankenkasse ist möglich. Eine digitale Pflegeanwendung hingegen können Pflegebedürftige ausschließlich bei Ihrer Pflegekasse beantragen.
Antrag des Endanwenders für eine DiPA – Ablauf
Die Versorgung mit einer DiPA wird von der Pflegekasse auf Antrag der pflegebedürftigen Person bewilligt. Die erste Bewilligung muss befristet werden und darf höchstens sechs Monate umfassen. Innerhalb dieser Frist prüft die Pflegekasse, ob die digitale Pflegeanwendung tatsächlich genutzt wurde und ob der Zweck der DiPA in der konkreten Versorgungssituation erreicht wird.
Ergibt diese Prüfung, dass beide Voraussetzungen erfüllt sind, ist eine unbefristete Bewilligung zu erteilen. Ein neuer Antrag ist dafür nicht erforderlich.
Die unbefristete Bewilligung bedeutet, dass die Pflegekasse die monatliche Erstattung bis zu 50 Euro dauerhaft gewährt, solange die gesetzlichen Anspruchsvoraussetzungen bestehen.
2.2 Eine DiPA muss kein Medizinprodukt sein
40a Digitale Pflegeanwendungen SGB XI:
“(1) Pflegebedürftige haben Anspruch auf Versorgung mit Anwendungen, die wesentlich auf digitalen Technologien beruhen”
33a Digitale Gesundheitsanwendungen SGB V:
“(1) Versicherte haben Anspruch auf Versorgung mit Medizinprodukten niedriger Risikoklasse, deren Hauptfunktion wesentlich auf digitalen Technologien beruht […].”
Dies sind die einleitenden Worte beider Paragrafen im Gesetzestext, welche sofort deutlich machen, dass es in puncto Anforderungen an die digitale Pflegeanwendung einen großen Unterschied gibt.
Während DiGA explizit als Medizinprodukte der Klasse I oder IIa definiert werden, ist bei DiPA nur von “Anwendungen” die Rede. Eine DiPA kann zwar, muss aber kein Medizinprodukt sein. Somit sind viele regulatorische Anforderungen, mit denen DiGA-Hersteller kämpfen, für viele DiPA (auf den ersten Blick) nicht relevant.
Das BfArM stellt allerdings vergleichbare Anforderungen auch an DiPA, die kein Medizinprodukt sind. Beispielsweise müssen Hersteller über ein Qualitätsmanagementsystem verfügen und Vorgaben an die Sicherheit und Funktionstauglichkeit der App erfüllen. Im aktuellen DiPA Leitfaden (Stand 11.10.2023) werden daher zahlreiche ISO/IEC-Normen für Medizinprodukte referenziert und an vielen Stellen auf die Medical Device Regulation (MDR) verwiesen. Somit unterscheiden sich die Anforderungen an DiPA in den meisten Fällen nicht maßgeblich, da sie an den Anforderungen für Medizinprodukte nach MDR orientiert sind.
Sofern es sich um ein Medizinprodukt handelt, muss dieses einer niedrigen Risikoklasse (I oder IIa) angehören.
2.3 Preisobergrenze
Während die Preisgrenze sich für DiGA nach den anderen Anwendungen in derselben Kategorie richtet, ist für digitale Pflegeanwendungen eine fixe Preisobergrenze vorgesehen. Diese besagt, dass maximal 50 € pro Monat für eine DiPA erstattet werden können (§40b SGB XI). Dies wurde in Fachkreisen zwar kontrovers diskutiert, aber das Gesetz spricht klare Worte. Grundsätzlich kann eine DiPA auch teurer sein, Mehrkosten sind aber dann vom Konsumenten der DiPA selbst zu tragen.
2.4 Kann eine DiPA auch eine DiGA sein?
Eine Anwendung kann grundsätzlich sowohl eine DiPA als auch eine DiGA sein. Nach aktuellem Stand gilt für Hersteller, dass beide Anträge für ein und dieselbe App eingereicht werden können. Zu beachten sind hierbei aber Abweichungen in den Anforderungen. Sofern eine Listung in beiden Verzeichnissen erreicht wird, hat der Leistungsträger des Konsumenten individuell zu prüfen, von welcher Kasse die Anwendung erstattet wird.
3. Leitfaden für die DiPAV
Wir haben die Inhalte der DiPAV in Form eines Leitfadens zusammengefasst, welcher die Antragstellung sowie die Anforderungen und deren Nachweis übersichtlich erläutert. Besonders wichtig ist der Nachweis des pflegerischen Nutzens, welcher ebenfalls erklärt wird.
3.1 Anforderungen an DiPA
Die Anforderungen sind im Gesetzestext auf einem abstrakten Niveau aufgelistet. Eine zentrale Hilfestellung liefern die Anlage-Dokumente 1 und 2 der DiPAV, sowie die Anlage 1 zur DiGAV, denn diese beinhalten Checklisten, mit denen Hersteller die einzelnen Anforderungen an ihrem Produkt prüfen können.
Folgende Anforderungen gelten für DiPAs:
- Sicherheit & Funktionstauglichkeit (vgl. DiPAV Anlage 1): Hierbei wird zwischen Medizinprodukten und Nicht-Medizinprodukten unterschieden. Medizinprodukte erfüllen die Anforderungen an Sicherheit & Funktionstauglichkeit durch die CE-Konformitätskennzeichung. Nicht-Medizinprodukte müssen die Anforderungen nach Anlage 1 der DiPAV erfüllen, welche jedoch sehr ähnlich zu den Medizinprodukt-Anforderungen für Risikoklasse I sind. Diese erfordert beispielsweise den Aufbau eines Qualitätsmanagementsystems für Medizinprodukte (z.B. nach ISO 13485), sowie eines Risikomanagementsystems).
- Datenschutz und Datensicherheit: Neben der DSGVO und dem allgemeinen Stand der Technik in der Datensicherheit sind die vom BSI vorgegebenen Anforderungen und die vom BfArM für DiGA und DiPA festgelegten Datenschutz-Prüfkriterien zu erfüllen. Außerdem sind alle Anforderungen aus der Anlage 1 zur DiGAV auch für DiPA zu erfüllen. Da einige Punkte davon aber nicht 1:1 übertragbar sind, liefert der DiPA-Leitfaden des BfArM weitere Informationen, wie mit diesen umzugehen ist.
- Anforderungen an die Qualität: (vgl. DiPAV Anlage 2)
- Interoperabilität: Hier gelten ähnliche Bestimmungen, wie für DiGA. Ein interoperabler Export von Daten muss sowohl in maschinenlesbarer, als auch menschenlesbarer/ausdruckbarer Form angeboten werden. Die DiPA muss es ebenfalls erlauben, von NutzerInnen verwendete Medizingeräte anzuschließen, sofern Daten daraus benötigt werden.
- Robustheit
- Verbraucherschutz
- Altersgerechte Nutzbarkeit, Nutzerfreundlichkeit und Barrierefreiheit
- Unterstützung der Pflegebedürftigen und der Nutzer
- Qualität der pflegebezogenen Inhalte
- Sicherheit der Pflegebedürftigen (Patientensicherheit)
- Pflegerischer Nutzen
Was ist ein pflegerischer Nutzen?
Der pflegerische Nutzen einer DiPA liegt vor, wenn durch Verwenden der Anwendung einer Beeinträchtigung der Selbständigkeit oder der Fähigkeiten der pflegebedürftigen Person entgegengewirkt wird. Auch das Fortschreiten der Pflegebedürftigkeit einer Person ist darin inbegriffen.
Zudem muss ein pflegerischer Nutzen in mindestens einem der folgenden Bereiche gegeben sein:
- Mobilität
- kognitive und kommunikative Fähigkeiten
- Verhaltensweisen und psychische Problemlagen
- Selbstversorgung
- Bewältigung von und selbständiger Umgang mit krankheits- oder therapiebedingten Anforderungen und Belastungen
- Gestaltung des Alltagslebens und sozialer Kontakte
→ Neben diesen Bereichen kann der pflegerische Nutzen auch im Bereich der Haushaltsführung gegeben sein.
Wichtig ist hierbei noch zu erwähnen, dass NutzerInnen der DiPA nicht zwangsläufig die pflegebedürftige Person selbst sein müssen. Auch wenn pflegende Personen durch die Anwendung dabei unterstützt werden, in einem der oben genannten Bereiche Hilfe zu leisten, liegt ein pflegerischer Nutzen vor.
Zulässige Studien zum Nachweis des pflegerischen Nutzens
Um den pflegerischen Nutzen nachzuweisen, müssen Hersteller vergleichende Studien vorlegen. Dies können auch retrospektive vergleichende Studien einschließlich retrospektiver Studien mit intraindividuellem Vergleich sein.
Alternativ dazu sind prospektive Studien, und speziell randomisierte, kontrollierte Studien aufgrund der hohe Aussagekraft empfehlenswert. Die Studien sollen in Deutschland durchgeführt werden. Ist dies nicht möglich, muss eine Übertragbarkeit auf den deutschen Versorgungskontext belegt werden.
Leider wurde der bisher einzige öffentlich bekannte Antrag auf Listung im DiPA-Verzeichnis 2024 eben genau aufgrund der Studie abgelehnt. Laut der Pressemitteilung von Lindera kritisierte das BfArM unter anderem, dass die vorgelegte Studie die Effekte der App nicht unabhängig von bereits bestehenden Pflegeleistungen isoliert belegen konnte.
3.2 Antrag
Ablauf des Antrags
Der Antrag für die Aufnahme in das DiPA-Verzeichnis wird vom Hersteller an das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) gestellt.
14 Tage nach dem Eingang der vollständigen Antragsunterlagen bestätigt das BfArM dem Antragsteller den Eingang der Unterlagen oder fordert fehlende Unterlagen an. Der Hersteller hat bis zu drei Monate Zeit, fehlende Unterlagen zu ergänzen. Liegen nach Ablauf der Frist keine vollständigen Antragsunterlagen vor, wird der Antrag abgelehnt.
Nachdem alle Unterlagen vollständig sind, hat das BfArM im Normalfall 3 Monate Zeit, den Antrag zu prüfen und zu einer Entscheidung bezüglich der Aufnahme ins DiPA-Verzeichnis zu kommen.
Inhalt des Antrags
Es ist wichtig anzumerken, dass digitale Pflegeanwendungen nur dauerhaft gelistet werden können. Eine Erprobungsphase gibt es nicht. Das bedeutet im Klartext, dass alle erforderlichen Unterlagen – auch der Nachweis über den pflegerischen Nutzen – bereits zum Zeitpunkt der Antragstellung vorliegen müssen. Der Antrag umfasst:
- Elektronischer Antrag über das Antragsportal beim BfArM
- Unterlagen zum Nachweis des pflegerischen Nutzens
- Unterlagen zum Nachweis der Erfüllung aller Anforderungen
- Ggf. weitere Unterlagen (z. B. Gebrauchsanweisung oder bei Medizinprodukten Konformitätsbewertung nach MDR bzw. MDD)
Im Antrag muss der Hersteller zudem folgende Angaben machen (nach §2 DiPAV):
- Hersteller und identifizierende Merkmale der DiPA
- Die Zweckbestimmung
- Zugehörige benannte Stelle (falls zutreffend)
- Von der benannten Stelle ausgestellte Zertifikate und der Konformitätserklärung des Herstellers (falls zutreffend)
- Gebrauchsanweisung
- Zweckbestimmung, Wirkungsweise, Inhalt und Nutzung der DiPA in einer allgemeinverständlichen Form
- Funktionen der DiPA
- An der Entwicklung der DiPA beteiligten Einrichtungen und Organisationen (falls zutreffend)
- Quellen für die in der DiPA umgesetzten pflegebezogenen Inhalte und Verfahren, insbesondere pflegerisch-medizinische Leitlinien und Expertenstandards, Lehrwerke und Studien
- Nachweis eines pflegerischen Nutzens einschließlich ergänzender Unterstützungsleistungen Dritter (nach “Nachweis des pflegerischen Nutzens” und “Studien zum Nachweis des pflegerischen Nutzens”) in einer einfachen und allgemeinverständlichen Kurzfassung
- Gruppe von Pflegebedürftigen, für die ein pflegerischer Nutzen nachgewiesen wurde
- Pflegerischer Nutzen, der für die angegebene Gruppe von Pflegebedürftigen und Nutzern nachgewiesen wurde
- Studie/n des Herstellers zum Nachweis des pflegerischen Nutzens einschließlich ergänzender Unterstützungsleistungen Dritter
- Die Erfüllung der Anforderungen und Vorgaben an eine DiPA einschließlich der jeweiligen Fragebögen
- Vorgesehene Nutzerrollen
- Qualitätsgesicherte Nutzung der DiPA im häuslichen Umfeld inklusive Ausschlusskriterien für die Nutzung
- Die für die Nutzung der DiPA erforderlichen und ergänzenden Unterstützungsleistungen Dritter nach Art, Inhalt, Umfang und Dauer, sofern zutreffend
- Standorte der Datenverarbeitung
- Kompatibilitätszusagen des Herstellers in Bezug auf unterstützte Plattformen und Geräte sowie erforderliches Zubehör und sonstige Produktbestandteile (falls zutreffend)
- Genutzte Standards und Profile sowie menschenlesbare Exportformate zur Herstellung von semantischer, syntaktischer und technischer Interoperabilität
- Deckungssumme der vom Hersteller für Personenschäden abgeschlossenen Haftpflichtversicherung
3.3 Beratung durch das BfArM
Das BfArM berät Hersteller sowohl vor als auch nach der Aufnahme ins DiPA-Verzeichnis. Die Themen, die in den Beratungsgesprächen behandelt werden, werden dabei maßgeblich von Ihnen als Hersteller bestimmt. Dabei ist es möglich, allgemeine Verständnisfragen zu den DiPA-Anforderungen zu klären, oder aber produktspezifische Unterlagen zu diskutieren.
Für Hersteller, deren DiPA bereits gelistet ist, können hingegen Fragen rund um die Änderung des Produkts behandelt werden.
Die Beratungsgebühr liegt hierbei zwischen 250 und 5.000 Euro (eine zugegeben sehr große Spannweite).
3.4 Meine DiPA ist gelistet! Was nun?
Nach einem positiven Bescheid des BfArM wird Ihre DiPA in das Verzeichnis aufgenommen. Herzlichen Glückwunsch – An diesem Punkt haben Sie einen großen Teil der Arbeit geschafft und können mit Ihrem Team feiern. Ganz am Ziel sind Sie allerdings noch nicht: Innerhalb von drei Monaten nach der Listung wird mit dem Spitzenverband Bund der Pflegekassen der Vergütungsbetrag für die DiPA verhandelt. Zudem sind technische und vertragliche Rahmenbedingungen für die Zurverfügungstellung der DiPA nach § 40a Absatz 4 SGB VI zu regeln.
3.5 Schiedsverfahren
Der Preis für die DiPA wird innerhalb der ersten drei Monate nach Aufnahme in das BfArM-Verzeichnis zwischen Hersteller und der Pflegekasse verhandelt. Erzielen diese beiden Parteien keine Einigung, legt das Schiedsgericht den Preis der DiPA fest. Details über den Ablauf des Schiedsverfahrens erfahren Sie im Abschnitt 8 (§ 30 – § 32) der DiPAV.
4. Weitere Wege in die Kostenerstattung durch Krankenkassen
Neben dem DiPA-Verfahren gibt es außerdem einige weitere Wege, die Software-Herstellern die Kostenerstattung im deutschen Gesundheitssystem ermöglichen. Dazu gehören unter anderem:
-
- Überblick über alle Wege in die Kostenerstattung: Zu unserem Whitepaper
- Kostenerstattung für Software im Krankenhaus: Zu unserem Whitepaper
- Digitale Gesundheitsanswendungen – DiGA-Verfahren: Zu unserem Leitfaden
- Selektivverträge mit Krankenkassen: Zu unserem Leitfaden
- Erstattung von Software im GKV-Hilfsmittelverzeichnis (HMV): Zu unserem Leitfaden
- Zentrale Prüfstelle Prävention (ZPP): Zu unserem Leitfaden
- NUB als Kostenerstattung für Software-Produkte im Krankenhaus: Zu unserem Leitfaden
Neben den genannten Wegen in die Erstattung lohnt auch der Blick auf öffentliche Fördermöglichkeiten, die den Weg dahin überbrücken können:
- Der Innovationsfonds: Zum Leitfaden
- Viele weitere Förderungen für Digital Health-Produkte: Zum Leitfaden
5. Fazit
Die Anforderungen an DiPA allesamt zu erfüllen, ist gar nicht so leicht. Vor allem die Vorlage der Studie/n als Wirksamkeitsnachweis zum Zeitpunkt der Erstbeantragung gestaltet sich für Hersteller als Herausforderung. Für die Zulassung von DiGA gibt es ein Fast-Track-Verfahren, welches Herstellern ermöglicht, den Nutzen einer Anwendung innerhalb eines Jahres in einer Studie zu belegen. Ein solches Verfahren ist aber noch nicht für die DiPA umgesetzt. Es bleibt abzuwarten, ob der Fast-Track-Mechanismus wie geplant mit dem BEEP-Gesetz zeitnah gesetzlich verankert wird.
Dieser Leitfaden wird regelmäßig überarbeitet, um den aktuellen Stand zu garantieren.
QuickBird Medical entwickelt Health Apps und Medical Software für Unternehmen und Forschungseinrichtungen auf Auftragsbasis. Falls Sie eine Medical App, DiGA oder DiPA planen und Unterstützung bei der Entwicklung benötigen, setzen Sie sich gerne mit uns in Verbindung und wir besprechen das Vorhaben unverbindlich.
The post DiPA-Leitfaden: digitale Anwendungen für die Pflege appeared first on QuickBird Medical.
]]>The post DiGA: Leitfaden zum Nachweis positiver Versorgungseffekte appeared first on QuickBird Medical.
]]>Deshalb erhalten DiGA auch erst Ihre Daseinsberechtigung, wenn ihr positiver Versorgungseffekt nachgewiesen werden konnte. Erst dann haben Sie eine Chance, dauerhaft ins Verzeichnis des BfArM aufgenommen zu werden. Am Ende soll Ihre DiGA schließlich einen echten Mehrwert für den Patienten liefern. Denn auch Krankenkassen wollen nur für Maßnahmen bezahlen, die einen Nutzen haben.
In diesem Artikel geben wir Ihnen einen detaillierten Leitfaden darüber, wie der Nachweis positiver Versorgungseffekte zu erfolgen hat.
Inhalt
- 1. Was ist ein positiver Versorgungseffekt?
- 2. Notwendige Angaben im Antrag
- 3. Studie zum Nachweis positiver Versorgungseffekte
- 4. Evaluationskonzept (bei vorläufiger Aufnahme)
- 5. Zusammenfassung
1. Was ist ein positiver Versorgungseffekt?
Ein positiver Versorgungseffekt kann viele Gesichter haben. Das merken wir beispielsweise, wenn wir uns einen typischen Behandlungsverlauf bei einer Depression ansehen:
- Niedergeschlagenheit und andere depressive Symptomatik
- Erstbesuch beim Hausarzt
- Stellung einer Verdachtsdiagnose
- Überweisung zu einem Psychotherapeuten
- Wartezeit von 6 Monaten (Quelle: BR24)
- Diagnostik & Diagnosestellung
- Ambulante Psychotherapie (20 – 25 Wochen)
- Rückfallprophylaxe
Schon in dieser sehr vereinfachten Darstellung wird deutlich, in welchen Bereichen die Versorgung von Patienten verbessert werden kann. Dazu gehört nicht nur die Optimierung der Therapie, sondern auch die Verbesserung von strukturellen Parametern, wie etwa eine Verkürzung der Wartezeit oder die Stärkung der Rolle des Patienten in der Therapie. Grundsätzlich unterscheidet die DiGA-Verordnung (DiGAV) beim positiven Versorgungseffekt zwischen zwei großen Kategorien: medizinischer Nutzen und patientenrelevante Struktur- und Verfahrensverbesserungen.
1.1 Medizinischer Nutzen
Im Grunde ist ein medizinischer Nutzen jede Form der Verbesserung von Diagnostik und Therapie. Konkret sind folgende vier Kategorien in der DiGAV als medizinischer Nutzen definiert:
- Verbesserung des Gesundheitszustands
- z.B. Rückgang von Symptomen
- Verkürzung der Krankheitsdauer
- z.B. Schnellere Rehabilitation nach einer OP
- Verlängerung des Überlebens
- z.B. Frühere Erkennung von Herzerkrankungen
- Verbesserung der Lebensqualität
- z.B. besserer Umgang mit einer Erkrankung
QuickBird Medical Tipp: Wie man in unserem DiGA-Verzeichnis-Analyzer sieht, gibt es aktuell (Nov 2025) keine im Verzeichnis gelistete DiGA, die nicht einen medizinischen Nutzen nachgewiesen hat. Eine DiGA mit einem reinen strukturellen Effekt (pSVV – siehe nächstes Kapitel) hat generell schlechte Chancen auf eine Listung im DiGA-Verzeichnis. Sie sollten sich auf den medizinischen Nutzen fokussieren, um Ihre Erfolgswahrscheinlichkeit zu maximieren. Mehr Infos finden Sie in unserem DiGA-Verzeichnis-Analyzer: Link
1.2 Patientenrelevante Struktur- und Verfahrensverbesserung (pSVV)
Wie eingangs erwähnt, muss eine DiGA nicht zwangsläufig bei der Therapie oder Diagnostik einer Erkrankung unterstützen, um einen positiven Versorgungseffekt zu erzielen. Auch organisatorische Verbesserungen, wie etwa die Verkürzung von Wartezeiten oder ein einfacherer Zugang zu medizinischen Leistungen wird nach DVG als positiver Versorgungseffekt gewertet. Der pSVV unterteilt sich gemäß DiGAV §8, Absatz 3 in folgende Aspekte:
- Koordination von Behandlungsabläufen
- Eine DiGA kann beispielsweise die Koordination zwischen dem Patienten und mehreren Leistungserbringern (z.B. Psychotherapeut und Neurologe) optimieren, um einen besser organisierten Therapieablauf zu ermöglichen.
- Ausrichtung der Behandlung an Leitlinien und anerkannten Standards
- Umsetzung von Patientenleitlinien in Apps, um Betroffenen zu verdeutlichen, wie sie selbst an der Behandlung mitwirken können. Eine App könnte beispielsweise gesundheitsrelevante Informationen anzeigen und regelmäßig an notwendige Arzttermine erinnern.
- Adhärenz
- Unterstützung bei der Umsetzung von Maßnahmen, die gemeinsam mit dem Arzt vereinbart wurden. Dadurch soll der Patient in seiner aktiven Rolle bestärkt werden.
- Erleichterung des Zugangs zur Versorgung
- DiGA können dazu beitragen, den Zugang zu medizinischer Versorgung zu verbessern und somit Barrieren, wie den Wohnort oder andere Faktoren kompensieren.
- Patientensicherheit
- Im Vordergrund steht hier die Vermeidung von Behandlungsfehlern. DiGA können diesen vorbeugen oder den Patienten dabei unterstützen, Fehler selbst zu erkennen.
- Gesundheitskompetenz
- Menschen fällt es oft schwer, gesundheitsrelevante Informationen zu finden und zu interpretieren. DiGA können dieses Wissen gezielt transferieren und Patienten ein größeres Verständnis für ihre Erkrankung und die Therapie ermöglichen.
- Patientensouveränität
- DiGA können Patienten darin bestärken, an den Entscheidungsprozessen in der Behandlung mitzuwirken und seine Erfahrungen miteinfließen zu lassen.
- Bewältigung krankheitsbedingter Schwierigkeiten im Alltag
- Hier geht es um die Unterstützung bei der Kompensation und der Reduktion von Unsicherheiten im Alltag. Eine Anwendung kann dafür sorgen, dass sich ein Patient frühzeitig auf ein Symptom einstellen kann oder ihn bei alltäglichen Tätigkeiten unterstützen.
- Reduzierung der therapiebedingten Aufwände und Belastungen der Patienten und ihrer Angehörigen
- Viele Therapien sind mit großen Aufwänden seitens der Patienten verbunden. Eine App kann hier beispielsweise helfen, indem sie Wartezeiten verkürzt oder sinnvoll überbrückt oder die physische Anwesenheit bei Terminen minimiert.
Sieht man sich die verschiedenen Bereiche an, merkt man schnell, dass es pSVV vor allem darum geht, die Rolle des Patienten in seiner eigenen Versorgung zu stärken.
Ökonomische Aspekte werden hier bewusst nicht berücksichtigt. Wenn Ihre DiGA den Patienten und Krankenkassen finanzielle Erleichterungen ermöglicht, ist das ein gutes Argument für Ihre Preisverhandlungen, gilt aber nicht als positiver Versorgungseffekt.
QuickBird Medical Tipp: Da eine DiGA als Medizinprodukt zertifiziert sein muss, muss sie laut MDR auch eine Form von diagnostischem oder therapeutischen Nutzen haben. Das hat zur Folge, dass manche Anwendungen, die nur einen pSVV bewirken, nicht als Medizinprodukt gelten und somit auch keine DiGA werden können.
Lesen Sie hier mehr über die Zweckbestimmung nach MDR und die Voraussetzungen für Medizinprodukte.
2. Notwendige Angaben im Antrag
Egal, ob Sie einen Antrag auf dauerhafte oder vorläufige Listung im DiGA-Verzeichnis stellen – einige Angaben sind für alle Hersteller obligatorisch.
2.1 Angabe der Patientengruppe
Sie müssen angeben, für welche Patientengruppe ein positiver Versorgungseffekt erzielt werden soll. Dafür ist ausschließlich eine Eingrenzung nach ICD-Codes zulässig. Jeder ICD-Code beschreibt eine Diagnose, wobei sowohl drei- als auch vierstellige Codes zulässig sind. Dreistellige Codes umfassen eine breitere Patientengruppe, wohingegen ein vierstelliger Code eine nähere Spezifikation dieser Patientengruppe darstellt.

Bedeutung von ICD-Codes
Beispiel:
- Dreistelliger Code:
- F32 = Depressive Episode
- Vierstelliger Code:
- F32.0 = Leichte depressive Episode
- F32.1 = Mittelgradige depressive Episode
Falls Ihre DiGA bei mehreren Indikationen einen positiven Versorgungseffekt nachweisen soll, müssen Sie für jede Patientengruppe einen Nachweis erbringen. Sie können den Nutzen aber auch gleichzeitig für mehrere Indikationen nachweisen, wenn er im Wesentlichen für diese vergleichbar ist. Dazu müssen Sie aber auch alle Indikationen in Ihre Studie mit einbeziehen. Die finale Bewertung, ob der angestrebte Versorgungseffekt dann auch für alle Gruppen vorliegt, obliegt dem BfArM.
Achtung: Ihre DiGA kann am Ende auch nur bei den Indikationen verschrieben werden, für die Sie auch einen Nutzennachweis erbringen können. Definieren Sie in Ihrem Antrag beispielsweise nur F32.1 als Patientengruppe, kann ein Arzt oder Psychotherapeut die DiGA nicht an Patienten verschreiben, denen ein F32.0 (leichte depressive Episode) attestiert wird.
2.2 Angabe des positiven Versorgungseffekts
Eine DiGA muss mindestens einen positiven Versorgungseffekt nachweisen können. Zentral ist hierbei, dass dieser auch konsistent mit der Zweckbestimmung, den Funktionen und Inhalten der DiGA und den veröffentlichten Aussagen (z. B. in Werbematerial) ist. Daher sollten Sie sich zeitgleich mit der Medizinprodukt-Zertifizierung bereits an den Nachweis des positiven Versorgungseffekts denken.
Eine Zweckbestimmung kann nicht mehr so einfach geändert werden. Achten Sie also darauf, dass diese nicht allzu spezifisch formuliert ist. Sie wollen ja später nicht das Potenzial Ihrer DiGA verschenken, wenn diese z.B. auch für andere Indikationen geeignet ist.
QuickBird Medical Tipp: Ein Nachweis mehrerer positiver Versorgungseffekte kann einen großen Mehrwert für die späteren Preisverhandlungen schaffen. Hier erfahren Sie mehr über die Preisverhandlung mit dem GKV-Spitzenverband.
3. Studie zum Nachweis positiver Versorgungseffekte
Der Nachweis eines positiven Versorgungseffekts liefert die Daseinsberechtigung für jede DiGA. Selbstverständlich handelt es sich daher auch um eines der wichtigsten Dokumente, das dem BfArM vorgelegt werden muss. Beim Studiendesign gibt es klare Vorgaben, welche wir in diesem Kapitel behandeln.
Der Durchführungsort der Studie muss Deutschland sein. Denkbar sind zwar auch Länder, in denen das Gesundheitssystem mit dem deutschen vergleichbar ist – dies birgt allerdings große Mehraufwände und Sie müssen genau begründen, warum hier eine Vergleichbarkeit vorliegt.
Zudem muss die Studie auch in einem öffentlichen Studienregister eingetragen werden. Das anerkannte Primärregister für Deutschland ist das Deutsche Register klinischer Studien (DRKS) beim BfArM.
12 Monate nach Einreichung der Studienergebnisse beim BfArM muss die Studie auch veröffentlicht werden. Dabei ist eine Veröffentlichung auf der Webseite ausreichend.
Wenn Sie für die Zertifizierung als Medizinprodukt nach MDR im Zuge der klinischen Bewertung eine Studie durchführen müssen, sollten Sie in Betracht ziehen, diese auch direkt auf die Anforderungen des BfArM abzustimmen. Wenn Sie dabei sauber vorgehen, können Sie mit einer Studie sowohl die klinische Prüfung nach MDR durchführen als auch den positiven Versorgungseffekt nachweisen.
QuickBird Medical Tipp: Buchen Sie unbedingt ein Beratungsgespräch beim BfArM, um Ihr Studiendesign bzw. Studiensynopse vorab zu besprechen. Auch wenn Sie von Qualität Ihrer geplanten Studie überzeugt sind, kann das BfArM hier eine andere Meinung haben. Am Ende zählt die Meinung des BfArM bei der Zulassung.
3.1 Zulässige Studientypen
Ein kurzes Experteninterview mit meinem Hausarzt sollte doch als Studie reichen, oder? Nein, so einfach geht das nicht.
Anwendungen im DiGA-Verzeichnis werden von den Krankenkassen finanziert und flächendeckend von Patienten genutzt. Das BfArM hat daher ein starkes Interesse daran, dass zu diesen Produkten auch aussagekräftige Erkenntnisse vorliegen. Deshalb vertrauen sie ausschließlich quantitativen Untersuchungen, wie es in der klinischen Forschung üblich ist. Als Mindestanforderung gilt, dass es sich um eine vergleichende Studie handeln muss. Nur so kann ein aussagekräftiger Nutzennachweis erbracht werden.
Einfach gesagt: Sie vergleichen Patienten, die Ihre DiGA nutzen mit Patienten, die Ihre DiGA nicht nutzen.
Achtung: Es ist nicht gesagt, dass die Patienten, die Ihre DiGA nicht nutzen, keine Behandlung bekommen! Die Kontrollgruppe muss nämlich an der Versorgungsrealität orientiert sein und eine für ihre Situation übliche Behandlung erhalten. Schließlich wollen Sie nachweisen, dass Ihre DiGA den aktuellen Zustand verbessern kann.
Auch vergleichende Studientypen lassen sich noch einmal untergliedern. Beispiele hierfür sind Kohortenstudien, nicht-randomisierte- und randomisierte kontrollierte Studien.
QuickBird Medical Tipp: Randomisierte kontrollierte Studien haben eine sehr hohe Aussagekraft im Vergleich zu vielen anderen vergleichenden Studientypen. Dadurch sind Ihre Ergebnisse weniger angreifbar. Offiziell bringt die Wahl eines aussagekräftigen Studientyps keine Vorteile vonseiten des BfArM. Aktuell hat aber fast jede dauerhaft gelistete DiGA im Verzeichnis ihre positiven Versorgungseffekte mit einer randomisierten kontrollierten Studie nachgewiesen (Stand 14.06.2021).

Vereinfachte Darstellung einer randomisierten kontrollierten Studie
Bei einer kontrollierten Studie werden die Teilnehmer in Gruppen unterteilt. Im einfachsten Fall handelt es sich dabei um eine Kontroll- und eine Studiengruppe. Es ist aber auch denkbar, mehrere Gruppen zu formen, um die Studienergebnisse besser einordnen zu können. Teilnehmer in der Studiengruppe werden in der Studie Ihre DiGA verwenden.
Wichtig ist, dass die Kontrollgruppe an der Versorgungsrealität orientiert ist. Wenn Menschen mit einer Indikation üblicherweise eine bestimmte Behandlung erhalten, sollten die Teilnehmer in Ihrer Kontrollgruppe diese Behandlung auch erhalten. Denn nur so ist eine realistische Nutzenbewertung möglich. Erhalten Menschen üblicherweise keine Behandlung (z.B. wegen Wartezeiten auf einen Therapieplatz), müssen auch Studienteilnehmer in der Kontrollgruppe keine Behandlung erhalten.
Beispiel: Ihre DiGA soll eine ambulante Psychotherapie bei Depression unterstützen, indem sie zusätzliche Empfehlungen liefert, die einen Patienten im Alltag unterstützen. Üblicherweise erhalten Patienten in diesem Szenario also eine ambulante Psychotherapie. Sie könnten Ihre Studienteilnehmer demnach in folgende Gruppen unterteilen:
- Gruppe 1: Patienten, die nur ambulante Psychotherapie erhalten
- Gruppe 2: Patienten, die ambulante Psychotherapie erhalten & zeitgleich die DiGA verwenden
Da es immer mehr DiGA gibt, die flächendeckend eingesetzt werden, ändert sich in manchen Bereichen auch die Versorgungsrealität. Ist die Behandlung mit einer anderen DiGA also bereits ein gängiges Vorgehen, müssen Sie Ihre DiGA in der Studie auch mit der bereits etablierten DiGA vergleichen.
Die Versorgungsrealität, die Sie in der Kontrollgruppe abbilden müssen, kann also drei Formen haben:
- Nichtbehandlung
- Behandlung ohne Einsatz bestehender DiGA
- Behandlung mit Einsatz bestehender DiGA
Ein zusätzliches Qualitätsmerkmal von Studien ist die Verblindung. Verblindung soll verhindern, dass Studienteilnehmer wissen, in welcher Gruppe sie sich befinden. Bei Medikamentenstudien erhält eine Studiengruppe dazu beispielsweise oft ein Placebo-Präparat, welches keine medizinische Wirkung hat. Bei der Untersuchung von Apps ist die Verblindung allerdings nur schwer umzusetzen.
3.2 Stichprobe
Die Stichprobe umfasst alle Teilnehmer, die Sie in Ihre Studie mit einbeziehen. All diese Teilnehmer müssen die Diagnose aufweisen, die Sie in ihrer Patientengruppe spezifizieren.
Ein großes Fragezeichen ist oft die Stichprobengröße. Im Zuge der Fallzahlplanung müssen Sie herausfinden und begründen, welche Stichprobengröße für die Studie optimal ist. Zu kleine Stichproben sind weniger repräsentativ und führen häufig dazu, dass existierende Effekte nicht erkannt werden. Zu große Stichproben sind für Sie als Hersteller natürlich teuer (Rekruitierung der Teilnehmer, Einschluss in die Studie etc.). Daher ist es wichtig hier eine möglichst präzise Prognose zu machen.
Die optimale Stichprobengröße wird im Rahmen einer statistischen Fallzahlplanung bestimmt. Dabei werden Annahmen über den erwarteten Effekt, die Streuung der Messwerte sowie das gewünschte Signifikanzniveau und die Power getroffen, um die notwendige Anzahl an Teilnehmern zuverlässig zu berechnen. Grundlage dafür sind mathematische Modelle und sofern verfügbar Parameter aus Pilotstudien oder der Literatur. Ein Statistiker übernimmt diese Planung üblicherweise.
Es ist auch möglich, für die Studie bereits existierende Daten zu verwenden (z.B. Abrechnungsdaten von Krankenkassen). Dadurch müssten Sie die Kontrollgruppe nicht selbst abbilden, ist allerdings oft mit erheblichen Aufwänden verbunden und erfordert gute Argumentation beim BfArM. Dabei müssen Sie sich außerdem auf die bereits gemessenen Endpunkte beschränken und sicherstellen, dass diese auch valide gemessen wurden. Sie sind also in der Freiheit eingeschränkt und haben keinen Einfluss auf die Qualität der Daten.
Auch ein Vorher-Nachher-Vergleich ist grundsätzlich zulässig. In diesem Fall erheben Sie Daten von denselben Personen zu unterschiedlichen Zeitpunkten – z.B. einmal vor Anwendung der DiGA unter herkömmlicher Behandlung und einmal nach Anwendung der DiGA. Doch auch hier muss die Behandlung zum ersten Messzeitpunkt der Versorgungsrealität entsprechen. Dieses Vorgehen ist hauptsächlich bei stabilen Zuständen sinnvoll, in denen eine herkömmliche Behandlung keine Verbesserung mehr erzielt (z.B. Diabetes).
3.3 Endpunkte
Die Endpunkte einer Studie beschreiben ihre angestrebten Ziele. Grundsätzlich können Sie beliebig viele Endpunkte definieren, der primäre Endpunkt wird aber der angestrebte positive Versorgungseffekt sein – z.B. Verbesserung der Lebensqualität. Die Nutzenbewertung kann über beliebig viele Endpunkte gemessen werden.
Je mehr Endpunkte Sie untersuchen und je mehr Nutzennachweise Sie erbringen können, desto vorteilhafter ist dies für die Preisverhandlungen. Bei der Definition von Endpunkten helfen Erkenntnisse aus der Literatur und der Pilotstudie (falls eine solche vorliegt). Je mehr primäre Endpunkte Sie definieren, desto teurer wird aber auch die Studie z. B. aufgrund einer größeren Stichprobengröße. Die meisten erfolgreichen DiGA beschränken sich auf genau einen primären Endpunkt.
Für die reine Aufnahme ins DiGA‑Verzeichnis ist der Nutzennachweis für einen patientenrelevanten Endpunkt ausreichend. Auch wenn sie nicht komplett deckungsgleich sein müssen, müssen Endpunkte konsistent mit der Zweckbestimmung der Anwendung sein.
Wichtig: Es zählen nur patientenrelevante Endpunkte. Endpunkte, die keinen Nutzen für den Patienten darstellen, spielen beim BfArM keine Rolle. Dazu zählen beispielsweise ökonomische Aspekte.
3.4 Hypothesen
Jede Studie muss konkrete Hypothesen untersuchen. Diese müssen so formuliert sein, dass sie entweder beleg- oder auch widerlegbar sind. Bei Hypothesen handelt es sich um klare Aussagen, die überprüft werden sollen. Idealerweise formulieren Sie für jeden Endpunkt, für den Sie einen Effekt nachweisen wollen, eine eigene Hypothese.
Beispiele:
- Patienten, die die DiGA verwenden, haben nach sechs Monaten eine höhere Lebensqualität als Patienten, die eine herkömmliche Behandlung erhalten.
- Unter Einsatz der DiGA verkürzt sich die Behandlungsdauer im Vergleich zu einer herkömmlichen Therapie.
3.5 Methode
Bei der Messung von Endpunkten müssen anerkannte Methoden eingesetzt werden. Zudem sind Endpunkte auch oft nur durch Selbstauskünfte der Studienteilnehmer messbar. Zum Beispiel gibt es kein Maßband, welches die Lebensqualität eines Menschen punktgenau messen kann. Die wichtigsten Methoden sind medizinische Messgeräte und psychologische Fragebögen.

Beispiele für Messinstrumente für einige positive Versorgungseffekte:
- Lebensqualität:
- SF-36/SF-12
- SWLS – Satisfaction with Life Scale
- EuroQoL (EQ)-5D
- Bewältigung der Schwierigkeiten im Alltag:
- SASS – Soziale Aktivität Selbstbeurteilungs-Skala
- GAF – Global Assessment of Functioning
- Patientensouveränität/Selbstwirksamkeit:
- ASKU – Allgemeine Selbstwirksamkeitserwartung
- SWOP-9 – Fragebogen zu Selbstwirksamkeit-Optimismus-Pessimismus
Wenn Sie eigene Messmethoden einsetzen, müssen diese erst validiert werden.
3.6 Minimal Clinically Important Difference (MCID)
Der Minimal Clinically Important Difference (MCID) beschreibt den kleinstmöglichen Unterschied in einem Endpunkt, der für Patienten tatsächlich eine spürbare und relevante Verbesserung darstellt. Er hilft dabei einzuschätzen, ob ein statistisch signifikanter Effekt auch klinisch bedeutsam ist. In der Praxis führt genau dieser Punkt häufig zu Diskussionen zwischen Herstellern und dem BfArM: Während Hersteller einen bestimmten Effekt als ausreichend relevant ansehen, kann das BfArM diesen als klinisch nicht bedeutsam einstufen. Die Definition eines angemessenen MCID ist daher ein zentraler Bestandteil der Studienplanung und sollte frühzeitig gut begründet werden.
4. Evaluationskonzept (bei vorläufiger Aufnahme)
Bei einer vorläufigen Aufnahme möchte das BfArM natürlich zumindest einen Anhaltspunkt dafür, warum Ihre DiGA einen positiven Versorgungseffekt erzielen kann und wie man plant, diesen nachzuweisen.
Dies wird in Form eines Evaluationskonzepts sichergestellt und hier gilt eine sorgfältige Herangehensweise. Das Konzept muss von einer herstellerunabhängigen Institution erstellt werden und den allgemeinen wissenschaftlichen Standards entsprechen. Wichtig ist, dass das Evaluationskonzept zum Nachweis des positiven Versorgungseffekts geeignet ist, denn sie müssen sich bei der Durchführung der Studie auch an dieses Konzept halten.
Ein guter Grundstein für das Evaluationskonzept ist die verpflichtende “systematische Nutzerdatenauswertung”. Diese erfolgt auf Basis von Daten, die mit der DiGA erhoben wurden. Auch wenn es nicht öffentlich vom BfArM erwähnt wird, ist darunter eine Pilotstudie zu verstehen.
Diese sollte nicht unterschätzt werden! Das bedeutet nämlich unter anderem, dass Sie ein Ethikvotum dafür brauchen. Dafür ist von einer Wartezeit von mehreren Wochen oder gar Monaten auszugehen. Auch bei der Nutzerdatenauswertung muss Ihre Stichprobe eine gewisse Größe erreichen, um eine gewisse Aussagekraft zu haben (z.B. 50 Personen).
Des weiteren ist eine Literaturrecherche ist für das Evaluationskonzept notwendig. Diese soll einen Überblick über den aktuellen Stand der Forschung in Ihrem Bereich geben und Anhaltspunkte dafür liefern, welche Endpunkte untersucht werden. Die Ergebnisse der Nutzerdatenauswertung sollen zeigen, dass die DiGA funktioniert und den angestrebten Effekt bewirken kann.
Untersuchen Sie in Ihrer Pilotstudie gerne mehrere Endpunkte. Dies hilft Ihnen bei der späteren Fallzahlplanung und Sie können dann entscheiden, welche Endpunkte Sie auch in der geplanten Studie untersuchen möchten.
Dem Evaluationskonzept sind auch noch weitere Dokumente beizulegen. Ein Dokument, auf welches das BfArM ein besonderes Auge wirft, ist das Studienprotokoll. Hier sind insbesondere logistische Aspekte der Studie definiert, wie beispielsweise die Finanzierung, das Datenmanagement und Verantwortlichkeiten. Manche Informationen doppeln sich im Evaluationskonzept und dem Studienprotokoll. Achten Sie darauf, dass auch im Studienprotokoll alle relevanten Informationen enthalten sind.
Auch ein statistischer Analyseplan ist obligatorisch. Dieser enthält unter anderem Informationen zu Randomisierung, Verblindung, Endpunkten, Umgang mit fehlenden Daten und statistischer Analyse.
QuickBird Medical Tipp: Vor Durchführung der Pilotstudie empfehlen wir einen Termin beim BfArM, um die Studiensynopse zu besprechen.
4.1 Fristen in der Erprobungsphase
Mit dem Bescheid über die vorläufige Aufnahme ins DiGA-Verzeichnis gibt es kaum eine Verschnaufpause. Denn dann starten die 12 Monate, in denen DiGA-Hersteller die Möglichkeit haben, die geforderten Studienergebnisse vorzulegen. In diesem Zeitraum muss die Studie gemäß dem Evaluationskonzept vollständig durchgeführt und ausgewertet werden.
Gesetzlich ist zwar ein Antrag auf die Verlängerung des Erprobungszeitraums möglich, dieser muss aber plausibel begründet werden und ist beim BfArM ungern gesehen. Grundsätzlich gilt: kann der Nachweis aus eigenem Verschulden heraus nicht erbracht werden (z. B. durch falsche Planung), wird der Antrag abgelehnt. Zulässig sind nur externe Faktoren, wie etwa die Corona-Pandemie, wodurch die Studie nicht nach Plan durchgeführt werden konnte.
Spekulieren Sie also nicht schon zu Beginn auf die Verlängerung dieses Zeitraums. Wenn dies dem BfArM auffällt, wird auch der Antrag auf Erprobung abgelehnt.
Eine Verlängerung muss spätestens drei Monate nach Ablauf der Frist beantragt werden. Sie sollten allerdings sofort mit dem BfArM Kontakt aufnehmen, sobald sich abzeichnet, dass Sie diese nicht einhalten können.
5. Zusammenfassung
Der Nachweis eines positiven Versorgungseffekts ist essenziell für die dauerhafte Listung im DiGA-Verzeichnis. Der positive Versorgungseffekt wird in zwei Kategorien unterteilt: medizinischer Nutzen und patientenrelevante Struktur- und Verfahrensverbesserung.
Der positive Versorgungseffekt muss im Antrag gemeinsam mit der Patientengruppe (ICD-Code) spezifiziert werden. Den angegebenen Nutzen muss jeder Hersteller für seine DiGA im Zuge einer empirischen Studie nachweisen. Das BfArM hat hohe Anforderungen an derartige Studien. So gilt beispielsweise als Mindestanforderung, dass es sich um eine vergleichende Studie handeln muss. Empfehlenswert sind hierbei insbesondere randomisierte kontrollierte Studien.
Anforderungen an die Studie im Überblick:
- Es muss mindestens eine vergleichende Studie sein
- Die DiGA muss mit der Versorgungsrealität verglichen werden
- Die Studie muss einen positiven Unterschied in einem patientenrelevanten Endpunkt nachweisen
- Zur Messung von Endpunkten müssen validierte Messinstrumente eingesetzt werden
- Der Durchführungsort der Studie muss Deutschland sein
- Die Studie muss in einem öffentlichen Register eingetragen werden
- Die Studienergebnisse müssen vollständig veröffentlicht werden
Anträgen zur vorläufigen Aufnahme ins DiGA-Verzeichnis ist ein Evaluationskonzept beizulegen, welches zumindest belegt, dass die DiGA funktioniert und es Anhaltspunkte für positive Versorgungseffekte gibt.
Inhalte des Evaluationskonzepts (bei vorläufiger Aufnahme):
- Genaues Vorgehen bei der Studie
- Systematische Nutzerdatenauswertung (Pilotstudie)
- Literaturrecherche
- Studienprotokoll
- Statistischer Analyseplan
QuickBird Medical übernimmt die Entwicklung und Regulatorik Ihrer DiGA und begleitet Sie bis zur Aufnahme ins BfArM-Verzeichnis. Kontaktieren Sie uns jederzeit, falls Sie Fragen haben, oder ein konkretes Projekt umsetzen möchten.
The post DiGA: Leitfaden zum Nachweis positiver Versorgungseffekte appeared first on QuickBird Medical.
]]>The post DiGA in Österreich: Zulassung digitaler Gesundheitsanwendungen (2026) appeared first on QuickBird Medical.
]]>In diesem Artikel geben wir Ihnen einen Überblick über:
- den geplanten Zulassungsprozess in Österreich
- den aktuellen Stand (2026) der Anforderungen an DiGA in Österreich
- den Zeitplan zur Einführung eines DiGA-Zulassungsprozesses in Österreich
Inhalte dieses Artikels
- 1. Hintergrund des DiGA-Konzepts
- 2. Ausgangssituation
- 3. Zulassung als Medizinprodukt vs. Erstattung durch Krankenkassen
- 4. Der Weg zur Erstattung für DiGA in Österreich
- 5. Bisherige Erstattungsmöglichkeiten in Österreich
- 6. DiGA-Modelle in anderen Ländern
- 7. Fazit
1. Hintergrund des DiGA-Konzepts
Deutschland hat 2020 als Pionier das Konzept einer digitalen Gesundheitsanwendung („DiGA“) eingeführt. DiGA sind auch als „Apps auf Rezept“ bekannt und können vom Arzt an Patienten verschrieben werden. Für sie wurde in Deutschland ein spezieller Zulassungsprozess geschaffen: der DiGA-Fast-Track. Digitale Produkte, die diesen Zulassungsprozess erfolgreich durchlaufen haben, werden von den Krankenkassen auf Rezept erstattet.
Länder wie Frankreich und Belgien folgten mit eigenen Zulassungs- und Erstattungsprozessen, die sich am deutschen Fast-Track Verfahren orientieren.
In Zukunft könnte das Konzept einer DiGA auch nach Österreich kommen – mehr dazu in den untenstehenden Kapiteln.
2. Ausgangssituation für digitale Gesundheitsanwendungen
Ausgelöst durch Deutschlands Einführung des DiGA-Fast-Tracks, verstärkte sich 2020 auch in Österreich die Diskussion zur Einführung eines Zulassungsprozesses für digitale Gesundheitsanwendungen (DiGA).
Seit Juni 2023 bildet der Digital Austria Act die Basis dieser Gespräche. Er zielt darauf ab, das österreichische Gesundheitssystem für digitale Innovationen zu öffnen und einen rechtlichen Rahmen für die Integration von DiGA zu schaffen.
Unter Punkt 7.4 ist dort aufgeführt:
„Die Verschreibung qualitätsgesicherter Digitaler Gesundheitsanwendungen soll in Zusammenarbeit mit der Sozialversicherung ermöglicht werden und die telemedizinische Versorgung ergänzen.“
Wie genau dieser Zulassungsprozess aussehen soll, ist noch nicht abschließend geklärt. Aufgrund des intensiven Austauschs mit deutschen Stakeholdern scheint es möglich, dass sich Österreich an ähnlichen Standards wie Deutschland orientieren wird. So sollen Qualität und Effektivität der erstattungsfähigen DiGA sichergestellt werden.
3. Zulassung als Medizinprodukt vs. Erstattung durch Krankenkassen
Im Zusammenhang mit digitalen Gesundheitsanwendungen ist es wichtig, zwischen zwei Zulassungsprozessen zu unterscheiden:
- Der Zulassung von (digitalen) Medizinprodukten nach Medical Device Regulation: Durch die Zulassung nach MDR sind Sie befugt, Ihr Medizinprodukt in Europa auf den Markt zu bringen. Dies bedeutet jedoch nicht, dass Sie auch Geld von den Krankenkassen für Ihr Produkt erhalten. Es geht hier erstmal nur um die Freigabe, dass Ihr Produkt sicher und leistungsfähig ist und somit grundsätzlich von Anwendern genutzt werden darf.
- Die Zulassung von digitalen Gesundheitsanwendungen (DiGA) für die Erstattung durch Krankenkassen: Wenn Sie Ihr digitales Produkt über die Gelder von Krankenkassen finanzieren möchten, müssen Sie sich mit länderspezifischen Regelungen beschäftigen. In Deutschland ist dies z.B. der DiGA-Zulassungsprozess beim BfArM, der es Ihnen ermöglicht, Ihr Produkt durch Krankenkassen erstatten zu lassen. Hierfür muss Ihr Produkt übrigens bereits als Medizinprodukt nach MDR zugelassen sein.
Ein digitales Medizinprodukt in Österreich auf den Markt zu bringen, ist aktuell durch die MDR bereits fest geregelt und ohne Probleme möglich. Ein standardisierter Prozess, wie Sie speziell ein digitales Gesundheitsprodukt in Österreich durch Krankenkassen erstatten lassen können, wird hingegen erst noch erarbeitet.
 Eine stark vereinfachte Abbildung der regulatorischen Abgrenzungen von DiGA in Österreich.
Eine stark vereinfachte Abbildung der regulatorischen Abgrenzungen von DiGA in Österreich.
4. Der Weg zur Erstattung für DiGA in Österreich
Zum jetzigen Zeitpunkt ist die genaue Ausgestaltung des Zulassungsprozesses noch weitestgehend unklar. Sowohl von Herstellerseite als auch von offizieller Seite wie der österreichischen Agentur für Gesundheit und Ernährungssicherheit (AGES) wird oft der deutsche DiGA-Fast-Track als Referenz angegeben. Es werden jedoch keine einzelnen Aspekte des deutschen Verfahrens hervorgehoben. Auch die Möglichkeit einer vorläufigen Zulassung ohne abgeschlossenen Wirksamkeitsnachweis wurde noch nicht abschließend geklärt.
Es gibt jedoch einige Anhaltspunkte, die mehr Aufschluss über die geplanten Anforderungen eines österreichischen DiGA-Zulassungsprozesses geben.
4.1 Anforderungen an digitale Gesundheitsanwendungen in Österreich
Konkrete Empfehlungen für Anforderungen an DiGA im österreichischen Gesundheitssystem hat das AIHTA (Austrian Institute for Health Technology Assessment) 2020 in einer ersten Studie veröffentlicht. Das AIHTA ist ein unabhängiges Institut zur wissenschaftlichen Entscheidungsunterstützung im Gesundheitswesen. Gesellschafter sind insbesondere die Länder Österreichs sowie der Dachverband der Sozialversicherungen.
Folgende Kriterien empfahl das AIHTA zur Qualitätsüberprüfung von DiGA:
- In einem ersten Schritt sollen DiGA auf Vorliegen einer CE-Kennzeichnung, sowie einer Einordnung nach Risikoklassen laut MDR überprüft werden.
- Die gemeldete Risikoklasse nach MDR soll anschließend dazu verwendet werden, die notwendigen Evidenznachweise, wie zum Beispiel die Durchführung von Studien, festzulegen. Zuständig dafür ist die AGES (Agentur für Gesundheit und Ernährungssicherheit).
- Eine Zulassung als erstattungsfähige DiGA soll unter Einbeziehung des Dachverbands der Sozialversicherungen ausgestellt werden. Die Kriterien sind dabei klinische Wirksamkeit und Sicherheit, zu erwartende Kosten im Gesundheitssystem, Datensicherheit und -schutz sowie Usability und Interoperabilität.
- Der Prozess soll laut Empfehlung des AIHTA pilotiert und dabei angepasst werden.

Viele der Kriterien des AIHTA wurden auch auf der LISAvienna Regulatory Konferenz im Herbst 2023 diskutiert:
- Therapietreue als Qualitätskriterium
- Nutzung von (anonymisierten) Gesundheitsdaten für ärztliches Fachpersonal, Hersteller oder zu Forschungszwecken
- Keine Brüche im digitalen Versorgungsweg: Anbindung an andere digitale Lösungen im Gesundheitswesen wie die österreichische Patientenakte ELGA
- Notwendigkeit der Bekanntmachung von digitalen Gesundheitsanwendungen, um Interesse an DiGA seitens Patienten sowie von ärztlicher Seite zu etablieren
Welche Kriterien schlussendlich im finalen Zulassungsprozess auch so übernommen werden, ist bisher noch nicht absehbar.
4.2 Zeitplan für DiGA in Österreich
Für die reguläre Erstattung von DiGA ist es von entscheidender Bedeutung, welche Anforderungen der Dachverband der Sozialversicherungsträger und die Österreichische Gesundheitskasse für DiGA festlegen. Erst danach ist absehbar, welche Art von digitalen Gesundheitsanwendungen überhaupt erstattungsfähig sind. Hier sollen die erarbeiteten Rahmenbedingungen im Rahmen der Pilotierung aus dem Jahr 2024 Aufschluss geben, die wir im nächsten Unterkapitel vorstellen.
Im Juni 2024 veröffentlichte das Bundesministerium für Soziales, Gesundheit, Pflege und Konsumentenschutz die „eHealth-Strategie Österreich“. Darin ist die konkrete Maßnahme zur Gestaltung eines Zulassungsprozesses für digitale Gesundheitsanwendungen festgelegt:
„M1.7: Schaffung eines einheitlichen Prozesses für die Bewertung digitaler Gesundheitsanwendungen (DiGAs) sowie digitaler Pflegeanwendungen (DiPAs) und in weiterer Folge bei darstellbarem Nutzen deren Einführung in die Gesundheitsversorgung. Priorisierung/zeitliche Planung: Kurzfristig (2024-2026)“
Das Pilotverfahren wurde November 2025 abgeschlossen. Bis Ende 2026 soll der gesetzliche Rahmen festgelegt werden, damit ab Anfang 2027 auch in Österreich DiGA verschrieben werden können.
4.3 Das Pilotierungsverfahren zur Schaffung eines österreichischen DiGA-Prozesses
Die Festlegung des Zulassungs- und Erstattungsverfahrens für DiGA wurde in Österreich im Rahmen eines Pilotprojekts vom Verband der Sozialversicherungen getestet. Die Praktikabilität des erarbeiteten Verfahrens sollte dabei anhand der Bewertung konkreter digitaler Gesundheitsanwendungen erfolgen.
Das Projekt wurde in einer schriftlichen Antwort an den SVDGV wie folgt skizziert:
| Initiator des Projekts | Dachverband der Sozialversicherungen |
| Ziel des Pilotverfahrens | Praxistest erarbeiteter Bewertungskriterien durch den Dachverband der Sozialversicherungen |
| Laufzeit | August 2024 bis Ende Februar 2025, danach Start der Umsetzungsphase |
| Kriterien an DiGA zur Aufnahme ins Verfahren | – DiGA ist Medizinprodukt der Klasse I oder IIa – Teilnahme unterliegt der Vertraulichkeit – Bereitschaft des DiGA-Herstellers an Meetings teilzunehmen und Rückfragen zu beantworten – keine Vergütung für die Teilnahme – keine Garantie für die tatsächliche spätere Vergütung teilnehmender DiGA |
| Definition des Projekterfolgs | Teilnehmende DiGA sollen das zuvor erarbeitete HTA-Framework durchlaufen haben. Es soll dadurch eine Bewertung möglich gewesen sein. Das erarbeitete Verfahren wird anschließend vorgestellt. |
| Einbezogene Stakeholder | e-Health Beauftragter der „Bundeskurie niedergelassene Ärzte“ (Keine Einbindung von Patienten oder Ärzten) |
| Vorteile für DiGA-Hersteller durch die Teilnahme | Einblick in den geplanten HTA-Prozess |
5. Bisherige Erstattungsmöglichkeiten in Österreich
Bereits jetzt können digitale Gesundheitsanwendungen in Österreich ausnahmsweise erstattet werden. Diese Anwendungen fallen unter die Kategorie „Sonstige notwendige Heilbehelfe“ gemäß § 137 des Allgemeinen Sozialversicherungsgesetzes (ASVG). Patienten können somit unter Einhaltung bestimmter Bedingungen einen Anspruch auf Kostenerstattung geltend machen.
Laut § 133 Abs. 2 ASVG ist es entscheidend, dass die DiGA den beabsichtigten Zweck erfüllt, ohne das notwendige Maß zu überschreiten. Die Überprüfung dieser Bedingung erfolgt individuell für jeden Antrag, wodurch die Erstattung nur in Einzelfällen gewährt wird.
Die Nutzung von Gesundheitsapps wird in Österreich bereits seit einiger Zeit gefördert. Eine Umfrage des österreichischen Dachverbands der Sozialversicherungen aus dem Jahr 2018 ergab, dass 9 von 16 Organisationen innerhalb der österreichischen Sozialversicherung insgesamt 22 Gesundheitsapps kostenfrei an ihre Versicherten abgaben. Auffällig dabei war, dass für die Mehrheit dieser Angebote kein Nachweis eines Versicherungsverhältnisses erforderlich war, was die Zugänglichkeit erhöhte. Im Jahr 2018 boten damit etwas mehr als die Hälfte der Krankenversicherungen in Österreich solche digitalen Gesundheitsanwendungen an.

Ein wesentliches Thema in diesem Zusammenhang ist die Qualitätsprüfung der angebotenen Apps. Bislang war die Klassifizierung als Medizinprodukt kein notwendiges Kriterium für die Bereitstellung der Apps. Es existiert hierzu keine einheitliche Qualitätskontrolle. Nichtsdestotrotz wurde versucht, bestimmte grundlegende Qualitätskriterien zu berücksichtigen, wie aus entsprechenden Grafiken und Auswertungen hervorgeht.

Interessanterweise konzentrierte sich der Großteil der verfügbaren Anwendungen auf die Prävention. Nur 5 der 22 untersuchten Apps waren in ein Behandlungs- oder Telemonitoring-Konzept integriert und somit speziell für erkrankte Personen konzipiert.
6. DiGA-Modelle in anderen Ländern
Immer mehr Länder planen einen standardisierten Prozess, um digitale Gesundheitsanwendungen in die Regelversorgung zu bringen. Neben Österreich sollten Sie außerdem einen Blick in die folgenden Länder werfen:
- Belgien (Link zum Leitfaden für die Zulassung von DiGA in Belgien)
- Frankreich (Link zum Leitfaden für die Zulassung von DiGA in Frankreich)
- Deutschland (Insider-Tipps für die Zulassung von DiGA in Deutschland)
- Italien (Link zum Whitepaper: Kommen DiGA bald nach Italien?)
- Schweiz (Link zum Leitfaden für die Erstattung von Digitalen Gesundheitsanwendungen (DiGA & dGA) in der Schweiz)
Neben standardisierten DiGA-Zulassungsprozessen gibt es außerdem die Möglichkeit, digitale Lösungen über Selektivverträge erstatten zu lassen. Mehr hierzu in unserem Whitepaper zu Selektivverträgen.
Außerdem existiert eine breite Palette öffentlichen Förderungen, welche sich für Digital Health-Produkte eignen: Link zu unserem Förderungsleitfaden für Digital Health-Produkte
7. Fazit
2025 scheint Bewegung in die österreichische Diskussion um die Einführung von digitalen Gesundheitsanwendungen (DiGA) gekommen zu sein. Trotz der großen geäußerten Motivation aller Beteiligten ist aktuell jedoch noch kein gesetzlicher Zulassungsprozess in Sichtweite.
Die Ergebnisse des AIHTA-Pilotierungsverfahrens werden gespannt erwartet. Der nächste wichtige Schritt ist die Festlegung der Anforderungen, die eine DiGA in Österreich erfüllen muss. Wichtig sind anschließend die konkreten Voraussetzungen für die Erstattung durch die Krankenkassen.
Erst dann bieten sich für Hersteller gesicherte Rahmenbedingungen, um DiGA in Österreich zu vertreiben.
Wir aktualisieren den Artikel bei neuen Entwicklungen. Abonnieren Sie unseren Newsletter, um hierzu auf dem neuesten Stand zu bleiben.
Der deutsche DiGA-Fast-Track – ein Modell für ganz Europa?
In diesem Whitepaper sehen wir uns an, in welchen EU-Ländern bereits ein DiGA-Modell existiert, ein DiGA-Modell geplant wird oder keinerlei DiGA-Modell in Aussicht steht. Außerdem klären wir die Frage: Kommt ein standardisiertes Modell in Europa für die EU-DiGA?
The post DiGA in Österreich: Zulassung digitaler Gesundheitsanwendungen (2026) appeared first on QuickBird Medical.
]]>The post Zertifizierung digitaler Kurse nach ZPP – Zentrale Prüfstelle Prävention appeared first on QuickBird Medical.
]]>Viele Software-Hersteller kennen die Erstattungsmöglichkeiten über den DiGA-Weg. Deutlich weniger bekannt ist der Monetarisierungsweg über die Zentrale Prüfstelle Prävention (ZPP). Die Zentrale Prüfstelle Prävention (ZPP) ermöglicht Anbietern, ihre digitalen Kurse (u.a. auch in Form von Apps) zertifizieren zu lassen, damit sie von Krankenkassen bezuschusst oder erstattet werden können. In diesem Artikel erfahren Sie, welche Voraussetzungen dafür gelten und wie der Weg zur erfolgreichen ZPP-Zertifizierung aussieht.
Auch rein digitale Angebote, ohne persönliche Kursleitung, können nämlich unter gewissen Umständen durch die Zentrale Prüfstelle Prävention (ZPP) zertifiziert und von den gesetzlichen Krankenkassen bezuschusst werden.
Überblick
- 1. Grundlage und Hintergrund
- 2. Was sind digitale Präventions- und Gesundheitsförderungsangebote nach Kapitel 7?
- 3. Formate digitaler Angebote
- 4. Abgrenzung zur DiGA (Sonderkapitel)
- 5. Die vier Handlungsfelder der Prävention und Gesundheitsförderung
- 6. Kriterien & Leitfaden zur Zertifizierung digitaler Präventions- und Gesundheitsförderungsangebote durch die ZPP
- 7. Nutzen und Wirtschaftlichkeit einer digitalen Präventionsanwendung
- 8. Weitere Wege in die Kostenerstattung durch Krankenkassen
- 9. Fazit
1. Grundlage und Hintergrund
Regulatorische Grundlage für die Arbeit der Zentralen Prüfstelle Prävention, die von allen gesetzlichen Krankenkassen gemeinsam getragen wird, ist der „Leitfaden Prävention“ des GKV-Spitzenverbandes. Historischer Hintergrund dessen war die Erkenntnis, dass das Gesundheitssystem sich zu einseitig auf rein kurative Maßnahmen stützt, und dabei die (Primär-)Prävention vernachlässigt.
Um digitale Präventionsangebote gezielt zu fördern und so einen Beitrag zur Gesundheitsförderung der Versicherten sowie zur Entlastung des Gesundheitssystems zu leisten, wurde der § 20 SGB V geschaffen. Er verpflichtet die Krankenkassen, Leistungen zur Primärprävention und Gesundheitsförderung zu unterstützen und entsprechende Angebote zu fördern. Auf Grundlage dieses Paragraphen können Krankenkassen die Kosten für zertifizierte Präventionskurse ganz oder teilweise übernehmen – sofern diese dem GKV-Leitfaden Prävention entsprechen und von der Zentralen Prüfstelle Prävention (ZPP) anerkannt sind.
Ursprünglich war dieser Pfad wohl eher für Kursangebote von z. B. Fitnessstudios und anderen Kursanbietern ausgelegt. Mit wachsendem Bewusstsein für die Bedeutung und Chancen der Digitalisierung im Gesundheitswesen entstand aber auch die Notwendigkeit, einen Rahmen für digitale Präventionsangebote zu schaffen.
Bereits zuvor konnten digitale Formate nach Kapitel 5 anerkannt werden, allerdings nur, wenn sie einer klassischen Kursstruktur mit einer realen Kursleitung folgten, etwa über Videokonferenzen oder Online-Termine. 2020 wurde daher, ein Jahr nach Einführung der DiGA, das Kapitel 7 in den Leitfaden Prävention aufgenommen. Es schafft erstmals die Grundlage für nahezu vollständig digitale, selbstgesteuerte Präventions- und Gesundheitsförderungsangebote, also Programme, die ohne direkte Kursleitung auskommen und eigenständig von den Versicherten genutzt werden können. Die Einweisung in die App und persönliche Unterstützung bei der Nutzung müssen jedoch weiterhin von qualifizierten Personen durchgeführt werden.
2. Was sind digitale Präventions- und Gesundheitsförderungsangebote nach Kapitel 7?
Digitale Präventions- und Gesundheitsförderungsangebote nach Kapitel 7 des GKV-Leitfadens unterscheiden sich inhaltlich und formal deutlich von den klassischen Kursen nach Kapitel 5. Während Kapitel 5 eine feste Kursstruktur mit aufeinander aufbauenden Modulen und klar vorgegebenem Ablauf vorsieht, ermöglicht Kapitel 7 eine flexible Gestaltung der Intervention. Anbieter können Inhalte, Ablauf und Nutzungslogik frei konzipieren, solange die Wirksamkeit und Qualität nachgewiesen sind.
Auch formal gelten eigene Anforderungen. Neben einem schlüssigen Interventions- und Evaluationskonzept müssen Nachweise zur Datensicherheit, zum Datenschutz und zur Qualitätssicherung erbracht werden, etwa durch eine ISO-Zertifizierung. Kapitel 7 schafft damit den Rahmen für moderne, evidenzbasierte und technologisch vielfältige Präventionsangebote, die eigenständig digital umgesetzt werden können.
Während z.B. DiGA allerdings oft völlig ohne menschliche Leitung auskommen, muss es auch für ansonsten digital konzipierte Präventionsangebote eine Möglichkeit zur persönlichen Unterstützung geben. Die mit der Unterstützung betrauten Personen müssen dafür qualifiziert sein und übernehmen sowohl die Einführung in die Nutzung des Angebots als auch die Beantwortung individueller Rückfragen.
3. Formate digitaler Angebote
Grundsätzlich werden die folgenden Formate digitaler Präventions- und Gesundheitsförderungsangebote im offiziellen Leitfaden unterschieden:
3.1 Internet-Interventionen
Internet-Interventionen, auch Online-Gesundheitstrainings genannt, sind meist verhaltensorientierte Trainingsprogramme, die speziell für die Nutzung über das Internet entwickelt wurden. Sie orientieren sich an bewährten Methoden aus klassischen Präsenztrainings und sind klar strukturiert aufgebaut. Teilnehmende werden dabei von einer Fachperson begleitet, die online Rückmeldungen gibt oder bei Fragen unterstützt. Diese Begleitung wird im Leitfaden als „E-Coaching“ bezeichnet. Typischerweise bestehen Internet-Interventionen aus vier bis zehn Einheiten, die in der Regel wöchentlich über einen Webbrowser am Computer oder Tablet durchgeführt werden.
3.2 Mobile Anwendungen
Mobile Anwendungen, also Gesundheits-Apps, verfolgen in der Regel ein Trainingsprinzip, das auf die regelmäßige und meist tägliche Ausführung eines bestimmten gesundheitsbezogenen Verhaltens ausgerichtet ist. Im Gegensatz zu Internet-Interventionen, die nach einer festgelegten Zahl an Einheiten abgeschlossen sind, zielen mobile Anwendungen stärker darauf ab, gesundes Verhalten dauerhaft in den Alltag der Nutzerinnen und Nutzer zu integrieren.
Häufig beinhalten sie Funktionen zur Beobachtung oder Messung des eigenen Verhaltens und seiner Auswirkungen, etwa über digitale Tagebücher oder Sensoren. Die erfassten Daten können genutzt werden, um persönliche Ziele zu definieren, motivierende Rückmeldungen zu geben oder Fortschritte zu belohnen.
Mobile Anwendungen werden in der Regel auf dem Smartphone eingesetzt. Eine feste Struktur in Form von abgeschlossenen Trainingseinheiten gibt es meist nicht, da die Nutzung kontinuierlich und alltagsbegleitend erfolgt.
3.3 Hybride Trainingskonzepte
Hybride Trainingskonzepte verbinden die meist breiter angelegten Internet-Interventionen mit ihren längeren Nutzungsphasen mit mobilen Anwendungen, die durch häufigere, kürzere Nutzungseinheiten gekennzeichnet sind. Dadurch lassen sich strukturierte Lerneinheiten und alltagsnahe Übungsphasen miteinander kombinieren.
Ein Beispiel ist ein digitales Stressmanagement-Training, das aus mehreren aufeinander aufbauenden Lern- und Übungseinheiten besteht, die wöchentlich genutzt werden. Ergänzend dazu kann eine mobile Anwendung eingesetzt werden, die mehrmals täglich kurze Entspannungsübungen ermöglicht.
4. Abgrenzung zur DiGA (Sonderkapitel)
Digitale Gesundheitsanwendungen (DiGA) zeichnen sich unter anderem dadurch aus, dass es eben keine reinen Präventionsangebote sind. Sie dienen der Therapie von Krankheiten. Von der ZPP zertifizierte Angebote sollen Erkrankungen hingegen vorbeugen.
Zudem sind DiGA zwangsläufig Medizinprodukte (der Risikoklasse I, IIa oder IIb) nach MDR. Präventionsangebote können zwar ebenfalls als Medizinprodukte zugelassen sein (siehe Abbildung), das ist aber keine Voraussetzung für die Zertifizierung.
Erfahren Sie mehr darüber in unserem Artikel zu dem Thema DiGA.
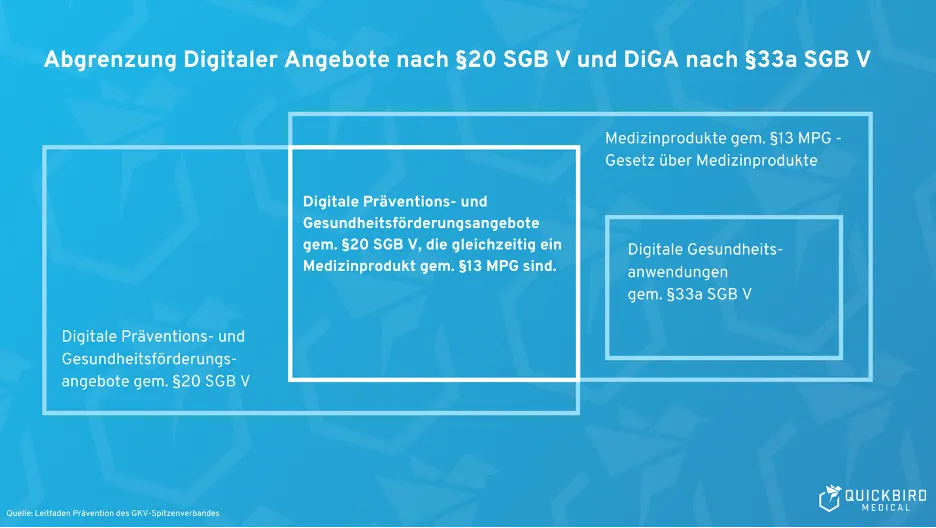
5. Die vier Handlungsfelder der Prävention und Gesundheitsförderung
Es gibt vier Handlungsfelder, innerhalb derer Angebote grundsätzlich zertifizier- und erstattbar sind:
5.1 Bewegungsgewohnheiten
Unter Bewegungsgewohnheiten zählen beispielsweise Maßnahmen, die Ausdauer, Beweglichkeit und Kraft fördern, Phasen der Inaktivität (z.B. Sitzen) verkürzen oder die Bewegungszeit verlängern. Auch Angebote zur Sturzprävention fallen in diese Kategorie. Allgemein geht es hier um alle Angebote, die auf die Steigerung physischer und/oder sportlicher Aktivität abzielen.
5.2 Ernährung
Angebote dieser Kategorie haben das Ziel, eine abwechslungsreiche und ausgewogene – also gesundheitsförderliche – Ernährung zu erzielen, und zum achtsamen Essen anzuregen. Zudem geht es darum, den Verzehr gesunder Lebensmittel zu fördern, und jenen ungesunder zu reduzieren.
5.3 Stress- und Ressourcenmanagement
Kurse in diesem Bereich zielen darauf ab, das allgemeine Stresserleben zu verändern, und Techniken zur Entspannung und zum Umgang mit Stress zu vermitteln. Das kann beispielsweise durch Achtsamkeitsübungen oder die Stärkung von Selbstwertgefühl und Selbstwirksamkeit erreicht werden. Auch Angebote zur Verbesserung der Schlafqualität fallen in diese Kategorie.
5.4 Suchtmittelkonsum
Im Handlungsfeld Suchtmittelkonsum geht es um Angebote zur Reduktion von z.B. Tabak- und Alkoholkonsum. Hier kann beispielsweise bei der Häufigkeit oder der Menge des Konsums angesetzt werden.
6. Kriterien & Leitfaden zur Zertifizierung digitaler Präventions- und Gesundheitsförderungsangebote durch die ZPP
Im Folgenden erläutern wir, welche Anforderungen Sie dafür erfüllen müssen und wie die Zertifizierung abläuft. Nähere Informationen hierzu finden Sie im Leitfaden Prävention unter Kapitel 7.
6.1 Gesundheitlicher Nutzen
Der gesundheitliche Nutzen stellt im Grunde die Daseinsberechtigung Ihres Produkts dar. Er leitet sich vom Handlungsfeld ab, in dem Ihr Angebot eingesetzt werden soll.
Ist Ihr Handlungsfeld definiert, gilt es auszuarbeiten, inwiefern Ihr Produkt hier einen Mehrwert liefern kann. Diesen Mehrwert müssen Sie anschließend durch eine Studie nachweisen (mehr hierzu weiter unten). Welche Endpunkte zulässig sind, erfahren Sie im Dokument Kriterien zur Zertifizierung digitaler Präventions- und Gesundheitsförderungangebote.
Einige Beispiele für Endpunkte sind:
- Steigerung der Bewegungszeit (Handlungsfeld Bewegungsgewohnheiten)
- Reduktion des Zuckerkonsums (Handlungsfeld Ernährung)
- Stärkung des Selbstwertgefühls (Handlungsfeld Stress- und Ressourcenmanagement)
- Reduktion der wöchentlichen Trinkmenge (Handlungsfeld Suchtmittelkonsum)
6.2 Anforderungen an die Studie
Ziel der Studie ist, einen Effekt Ihres Angebots über einen bestimmten Zeitraum hinweg nachzuweisen. Ein Vorteil: Der Vergleich mit einer Kontrollgruppe ist nicht notwendig (aber selbstredend nicht ausgeschlossen), stattdessen ist ein intraindividueller Vergleich ausreichend. Dazu werden die definierten Endpunkte zu vorgegebenen Zeitpunkten gemessen und miteinander verglichen, um die Verbesserung durch Ihr Angebot zu evaluieren.
Es besteht die Möglichkeit zur vorläufigen Zulassung vor dem vollständigen Nachweis des gesundheitlichen Nutzens für zunächst ein Jahr, sofern alle anderen Kriterien erfüllt werden.
Sofern die Studie erfolgreich war, wird Ihre Anwendung anschließend für drei Jahre durch die Zentrale Prüfstelle Prävention anerkannt.
6.3 Verfügbarkeit individueller Unterstützung
Auch wenn es sich bei Ihrem Angebot um eine Web-Anwendung oder eine mobile App handelt, ist persönliche Unterstützung und Erreichbarkeit Pflicht. Dies umfasst insbesondere den Nutzer-Support, welcher bei technischen, inhaltlichen und gesundheitlichen Themen unterstützt. Dabei kann es sich zum Beispiel um eine einzelne Person (z. B. Sie selbst) oder ein größeres Team handeln. Obligatorisch ist dabei, die Qualifikation der involvierten Personen sicherzustellen (z.B. durch entsprechenden Studienabschluss) und eine Einweisung in das Angebot dokumentiert zu geben.
6.4 Qualität
Ihr Angebot muss eine klar definierte Zielgruppe haben, und auch auf mögliche Kontraindikationen hinweisen. Alle inhaltlichen Aussagen, sowohl in der Anwendung selbst als auch in deren Marketing, müssen durch Quellen belegt sein. Zum Beispiel:
- “Regelmäßiges Ausdauertraining reduziert Ihr Herzinfarktrisiko.”
- “Progressive Muskelrelaxation trägt zu einer Reduktion des Stresserlebens bei.”
- “Der Konsum sozialer Medien vor dem Einschlafen verringert die Schlafqualität.”
Außerdem tragen Sie Verantwortung dafür, dass Nutzer verstehen, wie Ihr Angebot funktioniert. Beachten Sie, dass nicht nur Nutzer des Angebots, sondern auch etwaige eingebundene Personen (z.B. Coaches) durch entsprechende Anleitungen eingewiesen werden müssen.
6.5 Nutzerfreundlichkeit und Usability
Die Schnittstelle zwischen Mensch und digitaler Technik ist schon lange ein großes Thema. Ganze Forschungsfelder beschäftigen sich mit Fragen rund um die Gestaltung von Nutzeroberflächen. Auch bei digitalen Präventionsangeboten spielt diese eine zentrale Rolle. Die Ziele sind hier:
- Hohes Engagement der Nutzer
- Positives Nutzungserlebnis (User Experience)
Dies kann etwa durch den Einsatz von Gamification-Elementen, oder durch viele interaktive Inhalte erreicht werden.
Wichtige Quelle zur Umsetzung der Nutzerfreundlichkeit u.a. ist die Norm ISO 9241, auf die im Leitfaden des GKV explizit verwiesen wird.
6.6 Datenschutz & Datensicherheit
Im Bereich Datenschutz gilt es, die Vorgaben der DSGVO ebenso umzusetzen, wie die Vorgaben deutscher Datenschutzgesetze wie dem BDSG und SGB I. Entscheidend ist, ein Verständnis dafür zu entwickeln, welche Daten Ihre App verarbeitet und welche weiteren Parteien (Auftragsverarbeiter) eingebunden sind.
Abseits des Datenschutzes ist auch die Datensicherheit (bzw. Informationssicherheit) ein zentrales Thema, dem genügend Beachtung geschenkt werden sollte. Der GKV-SV fordert beispielsweise die Etablierung eines zertifizierten Informationssicherheitsmanagementsystems (ISMS) nach ISO 27001. Zusätzlich müssen Sie sich bei der Entwicklung des Produkts an die TR-03161 des BSI halten. Dabei handelt es sich um eine “Technische Richtlinie” für die Entwicklung von Anwendungen im Gesundheitswesen, die diverse Aspekte der Datensicherheit beschreibt.
6.7 Weitere gesetzliche Anforderungen
Die Identifikation und Umsetzung weiterer gesetzlicher Anforderungen sind ganz grundsätzlich Ihre Obliegenheit als Anbieter. Neben Datenschutzvorgaben und den Anforderungen des Leitfadens Prävention könnte dies zum Beispiel die Medical Device Regulation (MDR) sein. Dies ist der Fall, wenn es sich bei Ihrem Produkt um ein Medizinprodukt handelt. Eine Hilfestellung bei der Erörterung, ob Ihr Produkt der MDR unterliegt, ist unser Blogartikel zu diesem Thema.
6.8 Antrag und Zertifizierung
Nach Abschluss der Entwicklung steht der Zertifizierungsprozess an. Dieser wird über das Online-Portal der Zentralen Prüfstelle Prävention (ZPP) abgewickelt. Sobald Sie einen Account angelegt haben, tragen Sie Ihr Angebot als “Kurskonzept” ein. Die Prüfung dauert nach Angaben der ZPP maximal 10 Werktage und ist für Sie kostenfrei. Beachten Sie aber, dass die Prüfung Ihres Angebots und etwaig eingebundener Personen (Kursleiter) separat erfolgt. Mehrere Prüfprozesse können jedoch zeitgleich initiiert werden.
Der Zertifizierungsprozess kann auch nach einer Ablehnung wieder neu gestartet werden. Die Gründe einer Ablehnung sollten durch die ZPP kommuniziert werden, um Ihnen die notwendigen Anpassungen zu ermöglichen.
Nach erfolgreicher Zertifizierung wird Ihrem Kurs eine individuelle ID zugewiesen, und er im Verzeichnis der ZPP aufgenommen.
7. Weitere Wege in die Kostenerstattung durch Krankenkassen
Neben der Zentralen Prüfstelle Prävention gibt es außerdem einige weitere Wege, die Software-Herstellern die Kostenerstattung im deutschen Gesundheitssystem ermöglichen. Dazu gehören unter anderem:
- Überblick über alle Wege in die Kostenerstattung: Zu unserem Whitepaper
- Kostenerstattung für Software im Krankenhaus: Zu unserem Whitepaper
- Digitale Gesundheitsanswendungen – DiGA-Verfahren: Zu unserem Leitfaden
- Selektivverträge mit Krankenkassen: Zu unserem Leitfaden
- Erstattung von Software im GKV-Hilfsmittelverzeichnis (HMV): Zu unserem Leitfaden
- Digitale Pflegeanwendungen – DiPA-Verfahren: Zu unserem Leitfaden
- NUB als Kostenerstattung für Software-Produkte im Krankenhaus: Zu unserem Leitfaden
- Finanzierung über den Innovationsfonds: Zu unserem Leitfaden
- Öffentliche Förderungen für Digital Health-Produkte: Zu unserem Leitfaden
8. Fazit
Mit dem Rahmen für digitale Präventions- und Gesundheitsförderungsangebote hat der Gesetzgeber eine wichtige Lücke geschlossen, die die Konzeption der DiGA als rein kurative Anwendung offengelassen hat. Die Anforderungen an entsprechende Lösungen sind in angemessener Form gegenüber der DiGA entschärft, aber keinesfalls zu unterschätzen.
Dennoch kann ein alltagstaugliches digitales Kursangebot neben dem gesundheitlichen Nutzen für die Anwender auch für Kursanbieter ein interessantes Geschäftsmodell sein.
QuickBird Medical bietet eine Plattform für digitale Gesundheitskurse an, und entwickelt bei Bedarf individuelle Präventions-Software-Lösungen. Kontaktieren Sie uns gern, falls Sie eine App oder Web-Anwendung im Präventions-Bereich planen.
The post Zertifizierung digitaler Kurse nach ZPP – Zentrale Prüfstelle Prävention appeared first on QuickBird Medical.
]]>The post NUB als Kostenerstattung für Software-Produkte im Krankenhaus appeared first on QuickBird Medical.
]]>Digitale Innovationen wie KI-gestützte Diagnosesoftware oder klinische Entscheidungsunterstützungssysteme versprechen aber, die Patientenversorgung im Krankenhaus in naher Zukunft grundlegend zu verbessern.
In der Realität stoßen solche Lösungen häufig auf ein wesentliches Problem: die eingeschränkte Möglichkeit, Software-Innovationen im stationären Bereich zu monetarisieren.
Dabei eröffnet das sogenannte NUB-Verfahren Krankenhäusern die Möglichkeit, zeitlich befristet innovative Methoden zu finanzieren. NUB steht für „Neue Untersuchungs- und Behandlungsmethoden“ und bezeichnet ein Verfahren, das den Kliniken erlaubt, innovative Verfahren vorübergehend zu erstatten, bis die dauerhafte Aufnahme in das DRG-System erfolgt (mehr Informationen zum DRG-System finden Sie unten).
Dieser Artikel richtet sich an (zukünftige) Hersteller von Software-Lösungen für den stationären Bereich. Die zentrale Frage, die wir beantworten: Eignet sich der Weg über das NUB-Verfahren, um Ihre Software-Lösung zu monetarisieren?
Wir geben Ihnen einen Überblick über das System, welches die Kostenerstattung in Krankenhäusern dirigiert, und geben Ratschläge für den optimalen Weg zur Monetarisierung Ihres Software-Produkts.
Inhaltsverzeichnis
- 1. Kostenerstattung im stationären Bereich
- 2. Was sind NUB?
- 3. Finanzierung von Softwarelösungen als NUB: Ist das möglich?
- 4. Fazit
- 5. Ausblick: Alternative Finanzierungswege für digitale Innovationen
1. Kostenerstattung im stationären Bereich
Zuallererst möchten wir Ihnen eine kurze Einführung in das Finanzierungssystem der stationären Versorgung geben, damit auch das NUB-Verfahren für Sie klarer wird. Wir gehen hierbei der Verständlichkeit zuliebe nicht auf Details ein und vereinfachen bestimmte Sachverhalte bewusst.
Allgemein werden Krankenhäuser in Deutschland dual finanziert:
- Investitionskosten (z. B. Neubauten, Modernisierung, Geräte): Diese werden von den Bundesländern getragen. Die Länder entscheiden auch über Bau, Erweiterung und Schließung von Kliniken und müssen damit die flächendeckende Versorgung sicherstellen.
- Betriebskosten (Behandlungskosten für Patienten): Diese werden von den Krankenkassen übernommen. Die Finanzierung der Betriebskosten erfolgt über Fallpauschalen und ein Pflegebudget. Der Vergütung liegt das sogenannte DRG-System zugrunde. Das steht für Diagnosis Related Groups oder auf Deutsch: Diagnosebezogene Fallgruppen.
Im Fokus dieses Artikels stehen die Betriebskosten, da NUB-Vergütungen als Ergänzung zum DRG-System genau in diesem Bereich zum Einsatz kommen.
1.1 Kurzer Überblick zu DRG-Fallpauschalen
Das DRG-System („Diagnosis Related Groups“) ist das zentrale Vergütungssystem für stationäre Krankenhausleistungen (im somatischen Bereich).
Jede Behandlung wird dabei einer Fallpauschale (DRG) zugeordnet, die den typischen Aufwand für Diagnose, Schweregrad und erbrachte Leistungen abbildet. Auf Basis dieser Zuordnung und Verrechnung erhält das Krankenhaus dann einen festen Geldbetrag für jede Behandlung.
Die gesamte Liste der DRGs und assoziierten Fallpauschalen lässt sich in einem Online-Katalog einsehen.
Wesentliche Elemente des Systems sind:
- Bewertungsrelationen: Jede DRG ist mit einem Kostengewicht versehen, das die durchschnittliche Ressourcenbeanspruchung widerspiegelt.
- Landesbasisfallwert: Dieser wird jährlich zwischen Krankenhausgesellschaften und Krankenkassen auf Landesebene verhandelt und mit der Bewertungsrelation multipliziert, um den tatsächlichen Preis einer Behandlung zu bestimmen (vereinfacht gesprochen).
- Pflegebudget: Seit 2020 sind die Pflegepersonalkosten aus den DRGs ausgegliedert und werden über ein separates Budget vergütet.
1.2 Erlaubnis mit Verbotsvorbehalt
Im deutschen Krankenhaussektor gilt das Prinzip der „Erlaubnis mit Verbotsvorbehalt“.
Das bedeutet: Neue Methoden dürfen eingesetzt werden, solange der Gemeinsame Bundesausschuss (G-BA) die Methode nicht ausdrücklich ausgeschlossen hat.
Das unterscheidet sich vom „Erlaubnisvorbehalt“, wie er im ambulanten Gesundheitssektor gilt: Dort darf eine neue Methode erst dann angewendet werden, wenn der G-BA hierzu explizit ein positives Votum abgegeben hat.
In Deutschland ist der Einsatz neuer Methoden stationär also zunächst erlaubt. Das heißt aber nicht automatisch, dass eine Klinik Ihr innovatives Produkt auch tatsächlich einführen wird.

Erlaubnisvorbehalt (ambulant) vs. Verbotsvorbehalt (stationär)
Denn die Finanzierung im DRG-System stellt die eigentliche Hürde dar: Ihr neues Produkt wird vermutlich bisher nicht als Teil einer existierenden DRG finanziell abgedeckt sein.
Es bleibt Ihnen daher meist nur der Weg, die Kliniken jeweils einzeln davon zu überzeugen, dass Ihre Software-Lösung Kosteneinsparungs- oder Umsatzsteigerungspotenziale für die Einrichtung und medizinische Vorteile für die Patienten mitbringt. Die Klinik muss die Produktkosten dann aus dem existierenden Budget finanzieren und hoffen, dass sich dieses Investment am Ende lohnt.
In der Praxis führt das zu einer sogenannten „Innovationslücke“: Kliniken dürfen neue Methoden zwar anwenden, können sie aber oft nicht kostendeckend abrechnen. Hier kommen sogenannte NUB ins Spiel:
2. Was sind NUB?
NUB steht für „Neue Untersuchungs- und Behandlungsmethoden“. Damit sind Methoden gemeint, die mit Fallpauschalen und Zusatzentgelten im DRG-System noch nicht sachgerecht vergütet werden können. Vom Gesetzgeber wurde ein Verfahren geschaffen, das es einzelnen Krankenhäusern ermöglicht, zeitlich befristete Vergütungen für diese NUB zu erhalten („NUB-Entgelte“).
Möchte ein Krankenhaus eine solche neue Methode einsetzen, stellt es dazu jährlich einen Antrag beim Institut für das Entgeltsystem im Krankenhaus (InEK). Wird die Methode positiv bewertet, können krankenhausindividuelle Vergütungen ausgehandelt werden, bis eine reguläre Abbildung im DRG-Katalog erfolgt.
Für digitale Lösungen oder Softwareprodukte könnte NUB dann eine Rolle spielen, wenn sie Bestandteil einer neuen Methode sind. Das wäre z. B. dann der Fall, wenn Ihr digitales Tool ein innovatives Behandlungskonzept oder eine neuartige Untersuchungsmethode ermöglicht.
Das NUB-Verfahren und die Statusvergabe
(Quelle: Reimbursement Institute)
Das Besondere: Die Finanzierung von NUB funktioniert dabei nicht pauschal bundesweit, sondern ausschließlich für das Krankenhaus, das den Antrag gestellt hat. Außerdem besitzen die Entgelte zunächst nur eine Gültigkeitsdauer von einem Jahr.
Nach Ablauf der Frist ist es unter Umständen möglich, die NUB fest im DRG-System zu verankern.
So viel zur Theorie: Doch gelingt so ein NUB-Antrag für eine softwarebasierte Methode auch in der Praxis?
3. Finanzierung von Softwarelösungen als NUB: Ist das möglich?
QuickBird Medical hat sich in enger Kooperation mit dem Pharma-Experten Dr. Arndt Schmitz mit dieser Frage beschäftigt: Könnten NUB ein relevanter Weg in die Erstattung für Hersteller von Software-Produkten sein?
Dr. Arndt Schmitz verfügt über mehr als 20 Jahre Erfahrung in pharmazeutischer Forschung und Entwicklung. Er leitete Projekte zu klinischer Entwicklung, Biomarker-Validierung und digitalen Gesundheitslösungen und war zuletzt als Medical Software Product Lead tätig. In seinem MBA in Health Care Administration kam er zuerst mit dem Thema NUB in Kontakt. Heute ist er weiterhin im Digital Health-Bereich aktiv.
NUB als Erstattungsoption
Als Ergebnis der bisherigen Recherchen und Gespräche ergibt sich leider ein ernüchterndes Ergebnis in Bezug auf die NUB-Finanzierung von Software-Produkten. Der Weg scheint schwer bis unmöglich gangbar zu sein und allgemein ist dieser wohl für kaum ein Software-Produkt gut geeignet.
Das hat die folgenden Gründe:
3.1 Grund 1: Software als Investitionsgut und nicht als Betriebsgut
Im stationären Kontext unterscheidet der Gesetzgeber, wie oben skizziert, zwischen Erstattung von Betriebskosten und Investitionskosten.
Betriebskosten sind z. B. Verbrauchsgüter wie Medikamente oder Verbandsmaterial, die einmalig verwendet und dann verbraucht werden. Diese Kosten werden über das DRG-System getragen und für Innovationen, die noch nicht im DRG-System sind, ggf. indirekt auch über das NUB-Verfahren.
Investitionskosten umfassen z. B. die Anschaffung eines Gerätes, dessen Kosten einmalig anfallen und sich über viele Jahre amortisieren müssen. Das kann die Heizung sein, aber auch ein OP-Tisch. Investitionskosten werden nicht vom DRG-System getragen, sondern müssen über die Bundesländer finanziert werden.
Somit werden die Kosten Ihres Software-Produkts über das NUB-Verfahren auch nicht erstattet. Die Kosten von Software-Produkten müssen buchhalterisch und steuerrechtlich leider normalerweise als Investitionsausgaben betrachtet werden. Die genaue Definition von Investitionskosten lässt sich im Krankenhausfinanzierungsgesetz § 2 nachlesen.
3.2 Grund 2: Sind Software-Produkte bei NUB nicht erwünscht?
Nun könnte man ja naiv annehmen, dass man der Klassifizierung als Investitionsgut ausweichen kann, indem man das Geschäftsmodell des eigenen Software-Produkts anpasst. Statt hoher initialer Anschaffungskosten (Investition) stellt man auf ein Pay-Per-Use-Modell um, damit die Kosten tendenziell eher einen Betriebskosten-Charakter aufweisen. Das ließe sich leicht über Dongles bzw. softwaregestützte DRM (Digital Rights Management)-Ansätze realisieren.
Abgesehen davon, dass diese Argumentation in Bezug auf Investitions- vs. Betriebskosten angreifbar ist, funktioniert dieser Ansatz auch aus anderen Gründen nicht. Softwareprodukte genießen leider wenig Priorität in einem ohnehin unterfinanzierten stationären System. Daher würden auch Softwareprodukte, die per Systemarchitektur pro Fall berechnet werden, beim NUB-Antrag unserer Recherche nach abgelehnt werden.
Die Kosten des Software-Produkts konkurrieren mit Kosten z. B. für medikamentenbasierte Therapien. Das Geld, das in Software-Betriebskosten gesteckt wird, fehlt dann an anderen Stellen. Das scheint auf Basis von Gesprächen mit Experten in diesem Umfeld die vorherrschende Situation und Mentalität zu sein.
3.3 Grund 3: Das „U“ in NUB sollte kleingeschrieben werden
In Bezug auf Neue Untersuchungs- und Behandlungsmethoden (NUB) nehmen Untersuchungsmethoden einen deutlich kleineren Stellenwert ein. Der Großteil der stattgegebenen NUB-Anträge bezieht sich auf medikamentenbasierte Behandlungsmethoden. Die Gründe dafür sind vielfältig.
Software (z. B. zur KI-basierten Krebserkennung in der Radiologie) wird aber meist in den Bereich der Untersuchungsmethoden eingesetzt. Untersuchungsmethoden, unabhängig davon, ob es sich um eigenständige Software oder ein Hardware-Produkt handelt, erhalten nur einen Bruchteil der Finanzierung über das NUB-Verfahren. Dr. Arndt Schmitz fasst es so zusammen: Das „U“ in NUB sollte eigentlich sinnbildlich kleingeschrieben werden.
3.4 Grund 4: Empirische Daten belegen diese Theorien
Das Team von QuickBird Medical hat eine öffentlich verfügbare Datenbank des Reimbursement Institute (Einrichtung der RI Innovation GmbH) mit bisherigen NUB-Anträgen analysiert. Aus tausenden Anträgen haben wir die wenigen softwarebezogenen Anträge identifiziert. Auch wenn wir keine Information zu Vollständigkeit und Korrektheit dieser Daten haben, werfen sie doch ein Schlaglicht auf die Situation.
Das Ergebnis:
- Gesamtzahl der NUB-Software-Anträge: Von den insgesamt 2445 Anträgen (seit 2014) konnten wir lediglich 11 NUB-Anträge für dedizierte Software-Produkte identifizieren. Die wenigen identifizierten Software-bezogenen Anträge konzentrieren sich ausschließlich auf Bildgebungs- und 3D-Verfahren.
- Negativ bewertete NUB-Software-Anträge: Von den 11 identifizierten Software-bezogenen Anträgen wurden alle abgelehnt. Sie wurden vom InEK mit dem Status 2 oder 4 bewertet. Mehr Infos zur Zahlen-Kategorisierung finden Sie hier.
- Positiv bewertete NUB-Software-Anträge: Demnach gab es bei 2445 Anträgen keinen einzigen positiv bewerteten Software-bezogenen Antrag. Wenn Sie diesen Artikel lesen und einen relevanten Antrag kennen, den wir übersehen haben, kontaktieren Sie uns gern.
Diese Analyse untermauert die initiale Einschätzung, dass Software-Produkte kaum Chancen auf eine Kostenerstattung im Rahmen einer NUB haben. Es gibt augenscheinlich keinen Fall, der jemals positiv bewertet wurde. Daher ist auch anzunehmen, dass zukünftige NUB-Anträge für Apps oder andere Software abgelehnt werden.
4. Fazit
Diese Analyse von QuickBird Medical und Dr. Arndt Schmitz zeigt deutlich: Das NUB-Verfahren ist kein erfolgversprechender Weg, um Softwareprodukte in die befristete oder dauerhafte Erstattung im Rahmen des DRG-Systems zu bringen. Bislang gibt es unseres Wissens nach keinen dokumentierten Fall einer erfolgreichen NUB-Erstattung für Standalone-Software (schicken Sie uns gern eine E-Mail, falls Sie von einem Fall erfahren). Die strukturellen Hürden sprechen dagegen, dass sich dies in absehbarer Zeit ändern wird.
Auch wenn dieses Ergebnis ernüchternd wirkt, ist es für Hersteller von Softwarelösungen dennoch wertvoll: Wer frühzeitig weiß, dass der NUB-Pfad für reine Software-Produkte faktisch verschlossen ist, kann dies in der Geschäftsmodell- und Market-Access-Planung berücksichtigen und Fehlinvestitionen vermeiden. Außerdem wird im Gespräch mit Fachkreisen irgendwann der Begriff „NUB“ fallen. Dann ist es auf jeden Fall gut, vorbereitet und informiert zu sein.
Der Krankenhaussektor bleibt trotzdem ein relevanter Markt. Softwareprodukte können auch ohne NUB erfolgreich in Kliniken eingeführt werden. Das läuft dann allerdings über das Investitionsbudget der Häuser. Hier kommt es auf überzeugende Argumente an: Senkung von Kosten, Effizienzsteigerungen, Umsatzpotenziale für bestimmte Leistungsbereiche und medizinische Vorteile für die Patienten. Gelingt es, Mehrwerte für jeden klar herauszustellen, können Krankenhäuser auch ohne Erstattungsregelungen bereit sein, in digitale Lösungen zu investieren.
Mit diesem Artikel war unser Ziel, Ihnen einen Überblick über das denkbare Instrument „NUB“ zu verschaffen, und dieses (der Natur eines Blogartikels entsprechend) vereinfacht einzuordnen. Vielen Dank an dieser Stelle auch nochmal an Dr. Arndt Schmitz für die hervorragende Kooperation in Bezug auf die Recherche und Verfassung dieses Fachartikels.
Falls Sie nach anderen Monetarisierungswegen für Ihre Software suchen, finden Sie im folgenden Kapitel eine Übersicht wichtiger Erstattungswege für Software-Produkte im deutschen Gesundheitswesen.
5. Ausblick: Alternative Finanzierungswege für digitale Innovationen
Neben dem NUB-Verfahren gibt es außerdem einige weitere Wege, die Software-Herstellern die Kostenerstattung im deutschen Gesundheitssystem ermöglichen.
Dazu gehören unter anderem:
- Überblick über alle Wege in die Kostenerstattung: Zu unserem Whitepaper
- Digitale Gesundheitsanwendungen – DiGA-Verfahren: Zu unserem Leitfaden
- Selektivverträge mit Krankenkassen: Zu unserem Leitfaden
- Zentrale Prüfstelle Prävention (ZPP): Zu unserem Leitfaden
- Digitale Pflegeanwendungen – DiPA-Verfahren: Zu unserem Leitfaden
- Finanzierung über den Innovationsfonds: Zu unserem Leitfaden
- Öffentliche Förderungen für Digital Health-Produkte: Zu unserem Leitfaden
The post NUB als Kostenerstattung für Software-Produkte im Krankenhaus appeared first on QuickBird Medical.
]]>The post Leitfaden: Ist Ihre Software ein Medizinprodukt? appeared first on QuickBird Medical.
]]>In diesem Artikel geben wir Ihnen detaillierte Hilfestellungen, damit Sie fundiert entscheiden können, ob Ihre App bzw. Software sich als MDR-Medizinprodukt qualifiziert.
Im Anschluss finden Sie hier einen Leitfaden zur Bestimmung der richtigen Risikoklasse für Ihre Software: Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden.
Update Juni 2025: Das offizielle Guidance-Dokument „MDCG 2019-11: Guidance on Qualification and Classification of Software […]“ wurde überarbeitet und in Revision 1 veröffentlicht. Dieser Artikel spiegelt alle Aktualisierungen wider. Eine genaue Auflistung steht im folgenden Kapitel: Zum Kapitel über Revision 1
Inhaltsverzeichnis
- 1. Terminologie: MDR, Medizinprodukt & CE
- 2. Motivation: Software als Medizinprodukt
- 3. Fokus des Artikels: MDR
- 4. Wann ist meine App/Software offiziell ein Medizinprodukt?
- 5. Die Zweckbestimmung entscheidet
- 6. Was bedeutet dies konkret?
- 7. Typen von Apps/Software als Medizinprodukt
- 8. Häufige Unklarheiten
- 9. Wer entscheidet schlussendlich?
- 10. Medizinprodukt-Zulassung ohne Regulatorik?
- 11. Juni 2025: Update der offiziellen MDCG-Guidance
- 12. Fazit
1. Terminologie: MDR, Medizinprodukt & CE
Im Rahmen dieses Artikels definiert sich der Begriff „Medizinprodukt“ über die Begriffsbestimmung in der Medical Device Regulation (MDR). Knapp zusammengefasst: Ein Medizinprodukt ist ein Produkt mit medizinischer Zweckbestimmung. Damit Medizinprodukte auf dem europäischen Markt in den Verkehr gebracht oder in Betrieb genommen werden können, müssen sie mit einer CE-Kennzeichnung versehen werden.
Eine Software als Medizinprodukt wird offiziell auch Medical Device Software (MDSW) genannt. Die Feststellung, ob eine Software unter die Definition eines Medizinprodukts fällt, wird als Qualifizierung bezeichnet.

Den gesamten Weg zur Medizinproduktzulassung (und somit zur CE-Kennzeichnung) beleuchten wir in diesem Artikel: Zulassung & Zertifizierung von Software-Medizinprodukten (MDR)
2. Motivation: Software als Medizinprodukt
Meist beginnt alles mit der Idee, eine App oder Software zu entwickeln, die im Gesundheits-/Medizinbereich Anwendung finden soll. Im zweiten Schritt stellt sich nun die Frage, ob Ihr geplantes Produkt ein Medizinprodukt ist oder nicht. Wir sind als Software-Dienstleister auf die Entwicklung von Medical Software spezialisiert und kennen die Problematik daher sehr gut. Diese Evaluation hat einen großen Einfluss auf die Zukunft Ihres Produkts. Je nachdem, ob Sie Ihre App als Medizinprodukt auf den Markt bringen, ändern sich viele Gegebenheiten.
Auf der Contra-Seite: eine App als Medizinprodukt…
- ist aufwändiger und damit kostenintensiver
- benötigt Fachwissen im Bereich Qualitätsmanagement, das man entweder intern aufbauen muss oder extern einkauft
Auf der Pro-Seite: eine App als Medizinprodukt…
- lässt sich ggf. (einfacher) von den Krankenversicherungen erstatten. Eine Klassifizierung als Medizinprodukt ist z.B. Grundvoraussetzung, um in das DiGA-Verzeichnis aufgenommen zu werden.
- hebt die eigene Anwendung von der Konkurrenz ab, die ggf. nicht als Medizinprodukt zugelassen wurde. Das CE-Kennzeichen schafft Vertrauen bei Patienten, Ärzten, Krankenkassen und sonstigen Healthcare-Professionals.
Die Entscheidung, ob die eigene App ein Medizinprodukt ist, liegt allerdings nur indirekt bei Ihnen. Ein gegebenes Produkt mit festgelegter Zweckbestimmung und Funktionalität fällt entweder unter die Definition eines Medizinprodukts oder nicht. Das hängt vom MDR-Gesetzestext ab. Daran müssen Sie sich also halten. Ansonsten handeln Sie nicht legal (dazu weiter unten mehr).
Um die Klassifizierung als Medizinprodukt zu vermeiden, können Sie aber natürlich die Zweckbestimmung, Funktionalität und Vermarktung des Produkts entsprechend verändern. Wenn Sie zum Beispiel erst vorhatten, eine App für die Unterstützung der Therapie von Reha-Patienten zu entwickeln, können Sie stattdessen eine Fitness-App entwickeln (die nicht speziell Patienten als Zielgruppe hat und bei der Therapie nicht Teil der Zweckbestimmung ist). Lesen Sie mehr in unserem Artikel zur Formulierung der Zweckbestimmung.
3. Fokus des Artikels: MDR
Wir konzentrieren uns in diesem Rahmen auf den europäischen Markt, und betrachten speziell die Medical Device Regulation (MDR). Da eine Zertifizierung als Medizinprodukt nach Medical Device Directive (MDD) seit dem 26.05.2021 nicht mehr möglich ist, gehen wir hier nicht weiter auf diese ein. Bei der Definition eines Medizinprodukts gibt es im Übrigen weitreichende Überschneidungen zwischen MDR und MDD.
Außerdem betrachten wir in diesem Artikel speziell eigenständige Software (auch Standalone Software genannt) als Medizinprodukt. Dazu zählen insbesondere auch mobile Apps. Wir gehen nicht weiter auf Hardware (wie Herzschrittmacher) ein.
4. Wann ist meine App/Software offiziell ein Medizinprodukt?
Bevor wir uns konkrete Beispiele ansehen, ist es wichtig zu verstehen, was „offiziell“ darüber entscheidet, ob Ihre App ein Medizinprodukt ist. Überraschenderweise hängt dies nur von einer einzigen Definition in der MDR ab. Ihre App ist ein Medizinprodukt, wenn diese unter die Definition („Begriffsbestimmung“) der MDR eines Medizinprodukts fällt:
„Medizinprodukt“ bezeichnet ein Instrument, einen Apparat, ein Gerät, eine Software, ein Implantat, ein Reagenz, ein Material oder einen anderen Gegenstand, das dem Hersteller zufolge für Menschen bestimmt ist und allein oder in Kombination einen oder mehrere der folgenden spezifischen medizinischen Zwecke erfüllen soll:
- Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten,
- Diagnose, Überwachung, Behandlung, Linderung von oder Kompensierung von Verletzungen oder Behinderungen,
- Untersuchung, Ersatz oder Veränderung der Anatomie oder eines physiologischen oder pathologischen Vorgangs oder Zustands,
- Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper — auch aus Organ-, Blut- und Gewebespenden — stammenden Proben
und dessen bestimmungsgemäße Hauptwirkung im oder am menschlichen Körper weder durch pharmakologische oder immunologische Mittel noch metabolisch erreicht wird, dessen Wirkungsweise aber durch solche Mittel unterstützt werden kann.Die folgenden Produkte gelten ebenfalls als Medizinprodukte:
- Produkte zur Empfängnisverhütung oder -förderung,
- Produkte, die speziell für die Reinigung, Desinfektion oder Sterilisation der in Artikel 1 Absatz 4 genannten Produkte und der in Absatz 1 dieses Spiegelstrichs genannten Produkte bestimmt sind.
5. Die Zweckbestimmung entscheidet
Konkret bedeutet dieser Paragraph u. a., dass für die Qualifizierung (die Entscheidung, ob Ihr Produkt ein Medizinprodukt ist), die Zweckbestimmung Ihres Produkts ausschlaggebend ist. Die Liste an Funktionen Ihrer App/Software ist nicht relevant bzw. hat nur indirekt Einfluss auf die Zweckbestimmung. Falls die Zweckbestimmung Ihrer App z. B. die Therapie, Diagnose oder Prävention von Krankheiten ist, muss sie als Medizinprodukt nach MDR klassifiziert werden und ist damit sogenannte Medical Device Software (MDSW). Der Begriff der „Zweckbestimmung“ wird von der MDR folgendermaßen definiert:
„Zweckbestimmung“ bezeichnet die Verwendung, für die ein Produkt entsprechend den Angaben des Herstellers auf der Kennzeichnung, in der Gebrauchsanweisung oder dem Werbe- oder Verkaufsmaterial bzw. den Werbe- oder Verkaufsangaben und seinen Angaben bei der klinischen Bewertung bestimmt ist;
Hier wird auch deutlich, dass die definierte Zweckbestimmung Ihres Produkts auch z. B. auf allen Marketing-/Verkaufs-Kanälen so kommuniziert werden muss. Sie können Ihr Medizinprodukt nicht unter einer anderen Zweckbestimmung vermarkten als Sie in der Konformitätserklärung angegeben haben.
Wenn Sie Hilfe bei der Formulierung der Zweckbestimmung brauchen, finden Sie in diesem Artikel weitere Informationen: Formulierung der Zweckbestimmung für (Software-)Medizinprodukte
Nehmen wir hierfür mal das Beispiel einer Diabetes-App. Falls Ihre Software die Zweckbestimmung „der Dokumentation von Glukosewerten“ hat, ist sie kein Medizinprodukt. Sie dient nicht der Therapie, Prävention, Diagnose usw. von Krankheiten. Sie ist ein reines Dokumentationstool ohne direkten medizinischen Zweck. Falls Ihre Anwendung jedoch die dokumentierten Werte auswertet und auf dieser Basis Ratschläge gibt („Sie sollten etwas essen“), liegen „Therapie-Empfehlungen“ vor. Die App hat somit z. B. den Zweck „der Therapie von Diabetes“. Sie ist ein Medizinprodukt.
Dass die Zweckbestimmung im Vordergrund steht, wird auch im Vorwort (19) der MDR nochmal verdeutlicht:
Es muss eindeutig festgelegt werden, dass Software als solche, wenn sie vom Hersteller speziell für einen oder mehrere der in der Definition von Medizinprodukten genannten medizinischen Zwecke bestimmt ist, als Medizinprodukt gilt, während Software für allgemeine Zwecke, auch wenn sie in Einrichtungen des Gesundheitswesens eingesetzt wird, sowie Software, die für Zwecke in den Bereichen Lebensstil und Wohlbefinden eingesetzt wird, kein Medizinprodukt ist. […]
Software/Apps für allgemeine Zwecke, und damit nicht die konkrete Anwendung im medizinischen Bereich, sind kein Medizinprodukt. Beispielsweise ist Microsoft Excel kein Medizinprodukt, weil es für Anwendungsfälle jeglicher Art bestimmt ist. Auch mit Excel lassen sich natürlich bestimmte Kennzahlen berechnen, die für die z.B. für die Diagnose einer Krankheit verwendet werden könnten. Dies ist jedoch nicht die Zweckbestimmung von Excel. Eine App/Software, die speziell die Berechnung von Kennzahlen zur Diagnose einer bestimmten Krankheit als Zweck hat, ist hingegen ein Medizinprodukt.
Hinweis: Es ist möglich, dass Ihre Software/App nicht unter die MDR, sondern die IVDR (In-vitro Diagnostics Regulation) fällt. Falls Ihre App für die Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben bestimmt ist, fällt sie unter die IVDR. Die Qualifikations-Kriterien sind ähnlich. Weitere Informationen zur IVDR-Softwareklassifizierung finden Sie in unserem Leitfaden: IVDR Software: Qualifizierung und Klassifizierung
6. Was bedeutet dies konkret?
Wie man oben sieht entscheidet ein einziger Artikel der MDR, welche Software oder Geräte ein Medizinprodukt sind. Sie müssen für diese Entscheidung also nicht die komplette MDR durchwälzen. Das scheint auf den ersten Blick ein Vorteil zu sein. Das ist jedoch leider eher ein Nachteil: die Begriffsbestimmung ist sehr knapp und damit sehr vage. Um in konkreten Fällen zu entscheiden, braucht es etwas mehr Hintergrundwissen und Guidance.
Daher gibt es Dokumente, die mit konkreten Beispielen versuchen den Sachverhalt besser zu erklären. Das wohl wichtigste Guidance-Dokument im Rahmen der MDR stammt von der MDCG (Medical Device Coordination Group) und trägt den Titel „MDCG 2019-11 Qualification and classification of software […]“. Innerhalb dieses Artikels beziehen wir uns auf dieses Dokument mit „MDCG Guidance“ (obwohl es eine Vielzahl an unterschiedlichen Guidance Dokumenten der MDCG gibt).
Die MDCG Guidance ist nicht rechtlich bindend, hilft aber bei der Interpretation des MDR-Paragraphen. Was wir in diesem Artikel mit Software als Medizinprodukt benennen, bezeichnet die MDCG Guidance als Medical Device Software (MDSW). Um die Entscheidung zu erleichtern, ob Ihre Anwendung eine MDSW ist (und damit ein Medizinprodukt), enthält die MDCG Guidance u.a. einen Entscheidungsbaum:
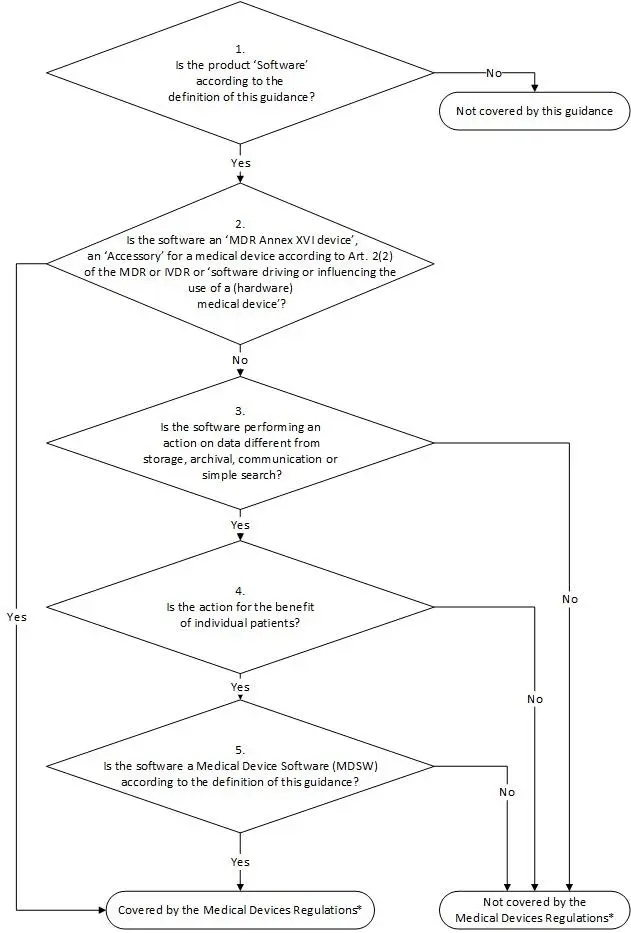
Entscheidungsbaum der MDCG Guidance: Zum Vergrößern klicken
7. Typen von Apps/Software als Medizinprodukt
Innerhalb der MDR und MDCG Guidance lässt sich zwischen drei verschiedenen Typen von Software/Apps als Medizinprodukt (bzw. MDSW) unterscheiden:
1. Zubehör eines Medizinprodukts
Falls Ihre Software/App ein anderes Medizinprodukt (meist Hardware) steuert oder beeinflusst, handelt es sich womöglich um Zubehör eines Medizinprodukts. Die MDR definiert dies folgendermaßen:
„Zubehör eines Medizinprodukts“ bezeichnet einen Gegenstand, der zwar an sich kein Medizinprodukt ist, aber vom Hersteller dazu bestimmt ist, zusammen mit einem oder mehreren bestimmten Medizinprodukten verwendet zu werden, und der speziell dessen/deren Verwendung gemäß seiner/ihrer Zweckbestimmung(en) ermöglicht […]
Die steuernde Software ist damit selbst auch ein Medizinprodukt.
2. Eigenständiges Medizinprodukt
Falls Ihre Software/App unabhängig eine eigene medizinische Zweckbestimmung hat, handelt es sich um ein eigenständiges Medizinprodukt. Eine App, die auf Basis bestimmter Eingabewerte Therapieempfehlungen gibt, ist z.B. ein eigenständiges Medizinprodukt. Der Großteil der DiGA wird z.B. in diese Kategorie fallen.
3. Teil eines Medizinprodukts
Bestimmte Software ist integraler Teil des Medizinprodukts. Die Software muss daher nicht eigenständig neu klassifiziert werden. Dazu zählt vor allem Embedded Software, wie z.B. die Software/Firmware eines Blutdruck-Messgeräts. Eine App wird kaum in diese Kategorie fallen, weil sie auf einem Smartphone läuft, welches selbst kein Medizinprodukt ist.
Je nach Typ gelten im Rahmen der MDR leicht abweichende Regelungen, auf die wir hier nicht näher eingehen werden.
8. Häufige Unklarheiten
Bei der Auswertung, ob ein Medizinprodukt vorliegt, gibt es einige typische Unklarheiten. Einen Teil davon lösen wir hier auf:
Kommunikation & Datenspeicherung:
Ein Chat oder eine Video-Call Lösung ist in der Regel kein Medizinprodukt. Der Zweck ist die reine Kommunikation. Dies gilt z.B. auch für reine Datenspeicherung/Dokumentation. Die MDCG Guidance schreibt hierzu ganz klar: „Storage, archival, communication & simple search is not medical product“.
Einfluss des Betriebssystems, der Laufzeitumgebung und „Aufenthaltsort“ der Software:
Ob Ihre Software auf einem entfernten Server läuft, ob Sie auf dem Smartphone läuft, ob sie auf Android oder Windows läuft, hat alles keinen Einfluss auf die Entscheidung, ob es ein Medizinprodukt ist. Wie oben beschrieben: die Zweckbestimmung zählt.
Mögliches Patientenrisiko:
Mit welchem Risiko Ihre App/Software einem Patienten Schaden zufügt, ist nicht relevant. Die Wahrscheinlichkeit und Schweregrad eines möglichen medizinischen Schadens haben keinen Einfluss darauf, ob Ihre App ein Medizinprodukt ist. Folgende Argumentation ist nicht zielführend: „Unsere App kann doch nur einen sehr geringen Schaden verursachen. Daher sollte es kein Medizinprodukt sein.“ Wenn der Zweck z.B. dennoch die Therapie einer Krankheit ist, reden wir weiter von einem Medizinprodukt.
Indirekter vs. direkter Schaden:
Oft wird angenommen, dass bestimmte Apps kein Medizinprodukt sind, weil sie „nur eine Entscheidungsunterstützung“ für den Arzt oder Patient darstellen. Am Ende entscheidet immer noch der Arzt bzw. Patient, wie gehandelt wird. Daher sollte es kein Medizinprodukt sein? Auch diese Argumentation ist nicht zielführend. Wenn Ihre App Therapieempfehlungen gibt, ist der medizinische Zweck die „Therapie einer Krankheit“ und damit ein Medizinprodukt. Und das ist auch richtig so: Ein überarbeiteter Arzt oder müder Patient vertraut tendenziell mehr und mehr seinem Software-Tool. Bestimmte Empfehlungen werden evtl. nicht mehr hinterfragt, und damit könnten potentiell falsche Entscheidungen mit schwerwiegenden Konsequenzen getroffen werden. Insofern sie für die Therapie von Diabetes bestimmt sind, sind also folgende Produkte damit gleichermaßen Medizinprodukte: eine physische Insulinpumpe, eine App zur Kontrolle einer Insulinpumpe, oder auch nur eine App, die konkrete Dosis-Empfehlungen für das Injizieren von Insulin gibt.
Nutzer der Software: Patient, Arzt oder ein „Laie“:
Nur weil Ihre App nicht von einem Arzt oder Patient benutzt wird, heißt es nicht, dass es kein Medizinprodukt ist. Eine Mutter könnte beispielweise Ihrem Kind mit eingeschränkter Mobilität bei der Therapie einer Krankheit helfen. Dafür verwendet die Mutter womöglich eine Software. Sie hat jedoch keine medizinische Ausbildung und ist nicht selbst ein Patient. Trotzdem ist der Zweck die Therapie. Damit besteht ein Risiko, dass die Therapie falsch durchgeführt, und damit ist die Anwendung auch moralisch gesehen zu Recht ein Medizinprodukt.
Vergleich mit anderen Apps am Markt
Gerne wird auch versucht über den Vergleich mit anderen Apps am Markt zu argumentieren: „App XY ist im App Store, hat die gleiche Funktionalität wie unsere App und ist kein Medizinprodukt. Wieso müssen wir dann ein Medizinprodukt sein?“
Auch der Vergleich mit anderen Apps hilft hier nicht weiter. Es gibt viele Apps am Markt, die wohl eigentlich als Medizinprodukt klassifiziert sein sollten, dies aber nicht sind. Daraus lassen sich jedoch keinerlei Schlüsse für die eigene App ziehen. Oft haben die Hersteller derartiger Apps aus reiner Unwissenheit agiert. Im Falle einer Klage/Mahnung, sind diese jedoch angreifbar. Wenn Sie ein stabiles Geschäft mit Ihrer App aufbauen wollen, sollten Sie sich an die Gesetzgebung halten.
9. Wer entscheidet schlussendlich?
Sie müssen selbst entscheiden, ob Ihre Software ein Medizinprodukt ist. Dazu können Sie das BfArM, Berater, Anwälte oder auch benannte Stellen hinzuziehen. Am Ende liegt die Entscheidung jedoch weiterhin bei Ihnen. Im Falle einer Klage, wird Ihre Entscheidung vom Gericht überprüft.
Falls Sie Ihre Software z. B. fälschlicherweise nicht als Medizinprodukt auf den Markt gebracht haben, tragen Sie die Konsequenzen. Die Überprüfung durch einen Richter, kann z. B. durch einen Schadensfall ausgelöst werden. Ein Patient oder die Konkurrenz könnte sie verklagen bzw. eine Abmahnung schicken.
Wenn Sie also unsicher sind, lohnt es sich, eine Einschätzung oder ein Gutachten von einer dritten Partei einzuholen. Folgende Möglichkeiten stehen Ihnen hierfür zur Verfügung:
- beim BfArM anfragen (die langen Antwortzeiten sind hier jedoch meist nicht ausreichend)
- ein Gutachten von einem spezialisierten Anwalt in diesem Bereich ausstellen lassen
- eine Einschätzung oder ein Gutachten von einem Regulatory-Berater einholen
Es ist nochmal wichtig zu betonen, dass auch diese Einschätzungen und Gutachten Sie nicht automatisch rechtlich schützen. Bei einer Klage, müsste Sie z. B. ein Anwalt immer noch vor Gericht auf Basis dieses Gutachtens verteidigen. Die Chance ist jedoch hoch, dass Sie den Prozess gewinnen, wenn Ihr Anwalt sorgfältige Arbeit geleistet hat.
10. Medizinprodukt-Zulassung ohne Regulatorik?
Gibt es einen Weg, die regulatorischen Pflichten für Medizinprodukt-Hersteller komplett zu vermeiden?
Ja, den gibt es: die Auslagerung der sogenannten Legalhersteller-Rolle an ein externes Unternehmen. Dies ist eine effiziente und risikominimierende Lösung, um Ihre Ressourcen zu schonen und gleichzeitig regulatorische Sicherheit zu schaffen. Sie müssen kein Qualitätsmanagement-System aufbauen und können den regulatorischen Zulassungsprozess vollständig abgeben.
So können Sie sich auf die Produktkonzeption und Vermarktung konzentrieren, während QuickBird Medical als Legalhersteller alle regulatorischen Anforderungen übernimmt und für die Einhaltung haftet.
Mehr Informationen dazu finden Sie hier: Zum Fachartikel
11. Juni 2025: Update der offiziellen MDCG-Guidance
Im Juni 2025 wurde ein in der Digital-Health-Szene lange erwartetes Update des offiziellen MDCG-Guidance-Dokuments zur Risikoklassifizierung und Qualifizierung durchgeführt.
Alle Änderungen wurden bereits in diesem Artikel berücksichtigt. Insgesamt hat sich in Bezug auf die Qualifizierung von Medical Device Software (MDSW) bzw. Medizinprodukt-Software nichts Grundlegendes geändert.
Die wichtigsten Änderungen waren die folgenden:
- Es wird mit mehr Details auf die Formulierung der Zweckbestimmung für MDSW eingegangen.
- Konkretisierung, dass die Kalkulation und Interpretation von medizinischen Informationen zu einer medizinischen Zweckbestimmung und Einstufung als Medizinprodukt führen könnte.
- Konkretisierung, dass Fitness- und Wellness-Apps keine Medizinprodukte darstellen.
- Neue Informationen bezüglich der Anwendbarkeit der European Health Data Space Regulation (EHDS)
- Eine breite Palette an anschaulichen Beispielen, die sich als Medizinprodukt oder Nicht-Medizinprodukt qualifizieren.
Die neue Version der MDCG-Guidance finden Sie hier.
12. Fazit
Es ist nicht immer einfach zu entscheiden, ob Ihre App/Software ein Medizinprodukt ist. Die Definition der MDR gibt nur sehr vage Vorgaben. Die MDCG-Guidance versucht mit anschaulichen Beispielen, ein besseres Verständnis zu vermitteln. Im Zweifelsfall ist es jedoch immer ratsam, nach einer dritten Meinung zu fragen. Falls Sie zusätzlich noch wissen möchten, ob Ihre App sich nicht nur als Medizinprodukt qualifiziert, sondern auch, in welche Risikoklasse diese fällt, lohnt sich ein Blick in unseren Artikel „Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden“.
Wir entwickeln Medical Apps und Medical Software auf Auftragsbasis für unsere Kunden. Daher sehen wir uns regelmäßig mit der behandelten Fragestellung konfrontiert. Für eine kostenlose Ersteinschätzung, ob Ihre App/Software ein Medizinprodukt ist, kontaktieren Sie uns jederzeit gerne.
The post Leitfaden: Ist Ihre Software ein Medizinprodukt? appeared first on QuickBird Medical.
]]>The post MDR Risikoklasse I vs. IIa: Unterschiede für Software-Medizinprodukte appeared first on QuickBird Medical.
]]>Die Risikoklassifizierung hat gravierende Auswirkungen und beeinflusst den Kosten- und Zeitrahmen der Produktentwicklung maßgeblich. Damit entscheidet die Risikoklassifizierung im Extremfall auch über den Erfolg oder Misserfolg eines jungen Unternehmens.
Die Klassifizierung erfolgt nach MDR basierend auf der Zweckbestimmung. Doch gerade bei Software führt diese Regelung oft zu Verwirrung und birgt Interpretationsspielraum – vor allem, wenn es darum geht, ob ein Produkt in die Risikoklasse I oder IIa fällt.
Hersteller stehen daher vor folgenden Fragen:
- Wie soll ich mit diesem Interpretationsspielraum bei der Klassifizierung von Medizinprodukten umgehen?
- Was sind die Konsequenzen dieser Entscheidung?
- Ist Risikoklasse II wirklich so viel schlimmer als Risikoklasse I? Was sind die konkreten Unterschiede für Sie als Hersteller?
In diesem Artikel liefern wir möglichst konkrete Antworten und helfen Ihnen die Konsequenzen der Klassifizierungs-Entscheidung besser zu verstehen.
Inhaltsverzeichnis
- 1. Die Risikoklassen nach MDR
- 2. Schwammige Klassifizierungsregeln bei Software-Medizinprodukten
- 3. Unterschiede: Risikoklasse IIa vs. Risikoklasse I
- 4. Unternehmerische Risiken: Risikoklasse I im Vergleich zur Risikoklasse IIa
- 5. Fazit
1. Die Risikoklassen nach MDR
Die MDR unterscheidet im Kern die folgenden Risikoklassen: I, IIa, IIb und III.
Der Sinn dahinter ist, dass mit steigendem Produktrisiko auch die regulatorischen Anforderungen strenger werden (risikobasierter Ansatz). Sie finden unter dem folgenden Link einen ausführlichen Leitfaden zur Bestimmung der Risikoklasse für Software-Medizinprodukte: Zum Leitfaden: Risikoklassifizierung von Software-Medizinprodukten
Im vorliegenden Artikel geht es vor allem um die Unterschiede zwischen Klasse I und IIa.
Hinweis: Wir verwenden die Schreibweise IIa statt 2a und I statt 1, da diese auch so innerhalb der MDR verwendet wird.
2. Schwammige Klassifizierungsregeln bei Software-Medizinprodukten
Wie oben bereits beschrieben, soll nach MDR vor allem das Risiko, das von einem Produkt ausgeht, über dessen Risikoklasse bestimmen – das lässt sich ja bereits vom Begriff ableiten.
Man könnte nun meinen, dass Software, von der kein nennenswertes Risiko ausgeht, in Risikoklasse I fällt. Aber nein: Diese pauschale Aussage ist leider nicht zulässig.
Bei der Bestimmung der Risikoklasse von (Standalone-)Softwareprodukten ist vor allem die berühmte Regel 11 der MDR relevant – und die führt bei vielen Herstellern zu Kopfzerbrechen.
Kurzzusammenfassung der Regel 11 nach MDR
Da wir die Regel 11 der MDR in einen anderen Artikel bereits umfassend behandelt haben, finden Sie hier nur die kurze Zusammenfassung jener Aspekte, die für diesen Artikel relevant sind.
Nach Regel 11 fallen alle Medizinprodukte, die einen der folgenden Zwecke erfüllen, mindestens in Risikoklasse IIa:
- Software, die dazu bestimmt ist, Informationen zu liefern, die für therapeutische oder diagnostische Zwecke herangezogen werden und
- Software, die für die Überwachung von physiologischen Prozessen bestimmt ist
Die Unterscheidung zwischen Risikoklasse I und IIa ist nach Regel 11 also nicht wirklich risikobasiert – lediglich der Verwendungszweck ist entscheidend. An dieser Stelle spielt weder der Schweregrad noch die Wahrscheinlichkeit eines möglichen Schadens eine Rolle.
Aber was sind „therapeutische oder diagnostische Zwecke“? Welche Software-Produkte fallen denn noch in Klasse I? Diese Frage haben wir in diesem Artikel umfassend behandelt: Zum Artikel: Welche Produkte fallen aktuell noch in Risikoklasse I?
3. Unterschiede: Risikoklasse IIa vs. Risikoklasse I
Wie weiter oben bereits angesprochen, macht es einen enormen Unterschied, ob Sie ein Medizinprodukt der Klasse I oder IIa zulassen. Hier beschreiben wir Aspekte, die für Sie als Hersteller besonders relevant sind:
- 1. Unterschied: Konformitätsbewertung zur Zulassung
- 2. Unterschied: Pflichten nach Zulassung
- a. Anzeigen von Änderungen
- b. Re-Auditierung
- c. Meldepflichten nach Markteintritt (PSUR)
3.1. Unterschied: Konformitätsbewertung zur Zulassung
Anforderungen: Risikoklasse I
Sie sind als Hersteller eines Klasse I Medizinprodukts natürlich dazu verpflichtet, die Vorgaben der MDR einzuhalten. Das umfasst beispielsweise den Aufbau eines Qualitätsmanagementsystems und die Erstellung der technischen Dokumentation für das Produkt. Der Vorteil ist aber, dass Sie die Konformität mit der MDR als Hersteller selbst – ohne Einbezug einer externen Partei – erklären können. Diese Option erwächst aus dem geringen Risikoprofil solcher Klasse I-Produkte.
Zusatzanforderungen: Risikoklasse IIa
Als Hersteller von Medizinprodukten der Klasse IIa (oder höher) sieht das Ganze anders aus. Sie sind dann darauf angewiesen, von einer externen und unabhängigen Organisation (benannte Stelle) auditiert zu werden. Erst wenn dieses Audit erfolgreich abgeschlossen ist, wird Ihnen die Konformität bestätigt und Sie können das Medizinprodukt auf den Markt bringen.
Auswirkungen auf den Zeitplan
- Wartezeit: Viele benannte Stellen sind kontinuierlich gut ausgelastet. Daher ist es ratsam, sich frühzeitig um einen Termin zu bemühen. Dennoch sollten Sie mit einigen Monaten Wartezeit rechnen, bevor die benannte Stelle mit dem Audit beginnt.
- Dauer des Audits: Es gibt keine gesetzliche Frist, wie lange ein Audit durch eine benannte Stelle dauern darf. Sie sollten mindestens 6 bis 12 Monate für diesen Audit-Prozess einplanen. Der Markteintritt Ihres Medizinprodukts verzögert sich somit maßgeblich, was gerade für Start-ups ein großes Problem darstellen kann.
- Aufbau des Qualitätsmanagementsystems: Ein Qualitätsmanagementsystem brauchen Sie zwar auch als Hersteller von Klasse I Produkten, allerdings ist der Anhang IX erst relevant, sobald eine benannte Stelle involviert ist. Dieser besagt unter anderem, dass die benannte Stelle ihr Qualitätsmanagement und auch dessen Konformität mit etwaigen harmonisierten Normen prüft (z.B. ISO 13485). Daher erwerben Hersteller i.d.R. an dieser Stelle ein ISO 13485-Zertifikat.
Auswirkungen auf die Kosten
- Audit-Kosten: Die Kosten für das Audit durch die benannte Stelle sind natürlich von Ihnen als Hersteller zu tragen. Diese belaufen sich unserer Erfahrung nach auf mindestens 50.000 € bis zu 200.000 €.
- Personalkosten: Das Audit der benannten Stelle muss auch von Ihrer Seite vorbereitet und begleitet werden. Die von der benannten Stelle gefundenen Abweichungen müssen von Ihnen als Hersteller behoben werden, wodurch Sie zusätzliche personelle und zeitliche Ressourcen einplanen müssen.
- Opportunitätskosten: Durch den Auditprozess mit der benannten Stelle verzögert sich Ihr Markteintritt um viele Monate oder sogar Jahre. Dementsprechend entgehen Ihnen für die Zeit auch Unternehmens-Umsätze. Auch dies kann gerade für Start-ups der ausschlaggebende Grund für eine Insolvenz werden.
3.2. Unterschied: Pflichten nach Zulassung
3.2.1. Unterschied: Anzeigen von Änderungen
Anforderungen an Risikoklasse I
Auch Hersteller von Medizinprodukten der Risikoklasse I müssen einem Change-Prozess folgen und jegliche Produktänderungen prüfen, freigeben und umsetzen. Dabei ist auch zu evaluieren, ob die Änderung Auswirkungen auf die Risikoklassifizierung hat.
Trotzdem sind Änderungen bei Software der Risikoklasse I ohne Involvierung einer externen Stelle möglich. Sie können Änderungen deutlich schneller veröffentlichen, weil Sie nicht auf den Ausgang einer externen Prüfung warten müssen.
Zusatzanforderungen an Risikoklasse IIa
Ab Risikoklasse IIa wird es mit Produktänderungen etwas komplizierter, weil auch hier die benannte Stelle einbezogen werden muss. Sogenannte „signifikante“ Änderungen müssen angezeigt werden und erfordern möglicherweise eine erneute Prüfung. Meldepflichtig sind beispielsweise Änderungen der Zweckbestimmung oder Anpassungen, welche die Leistungsfähigkeit des Produkts beeinflussen.
Auswirkungen der Risikoklasse IIa: Zeitplan
- Prüfung von Änderungen: kleinere Änderungen sollten auch ohne zusätzliche Prüfung möglich sein. Sobald die Änderungen am Produkt aber größer werden, sind Sie wieder von der Freigabe durch die benannte Stelle abhängig. Eine Prüfung kann in diesem Fall auch wieder einige Zeit in Anspruch nehmen, bevor sie die Änderung veröffentlichen können.
Auswirkungen der Risikoklasse IIa: Kosten
- Kosten für Prüfung: Die benannte Stelle wird Ihnen jegliche Prüfung in Rechnung stellen – genauso wie beim initialen Audit für die Marktzulassung. Je nachdem, wie umfangreich die Prüfung einer Produktänderung ausfällt, desto höher werden auch die finanziellen Aufwände.
- Verzögerter Release: Die Veröffentlichung von neuen Features kann auch geschäftlich relevant sein, wenn es sich beispielsweise um Funktionen handelt, die von vielen potenziellen Nutzern gebraucht werden. Auch in diesem Fall entgehen Ihnen womöglich Umsätze, solange die Prüfung der benannten Stelle noch andauert.
3.2.2. Unterschied: Re-Auditierung
Anforderungen an Risikoklasse I
Als Hersteller einer Software der Risikoklasse I werden Sie nicht regelmäßig auditiert. Die einzigen nennenswerten Prüfungen, auf die Sie sich einstellen müssen, sind sogenannte Herstellerüberwachungen durch die Aufsichtsbehörden. Diese können in unregelmäßigen Intervallen stattfinden. Die Aufsichtsbehörde wird dabei gezielt Dokumente von Ihnen anfordern und prüfen (z.B. den Risikomanagementprozess). Der Umfang und die Frequenz dieser Prüfungen sind aber in der Regel deutlich geringer als die Audits der benannten Stelle (bei Risikoklasse IIa und höher).
Zusatzanforderungen an Risikoklasse IIa
Hier verhält es sich etwas anders. Wir haben oben ja bereits von den Prüfungen durch die benannte Stelle (z.B. für die initiale Marktzulassung oder bei Produktänderungen) gesprochen. Doch auch, wenn Sie keinerlei Produktänderungen vornehmen, ist das Zertifikat der benannten Stelle nicht ewig gültig. Üblicherweise müssen Sie nach spätestens 5 Jahren erneut durch ein komplettes Audit und eine Prüfung der technischen Dokumentation der benannten Stelle. Ein Überwachungsaudit und eine stichprobenartige Prüfung der technischen Dokumentation finden zusätzlich jährlich statt.
Zusammengefasst haben Sie also verschiedene Prüfungen, die durchgeführt werden:
- MDR-Audit und Prüfung der technischen Dokumentation (spätestens alle 5 Jahre)
- MDR-Überwachungsaudit (jährlich)
- Herstellerüberwachung durch Aufsichtsbehörde (unregelmäßig)
Auswirkungen der Risikoklasse IIa: Zeitplan
- Die regelmäßigen Audits bzw. Prüfungen sollten keinen Einfluss auf den Zeitplan der Produktentwicklung haben.
Auswirkungen auf die Kosten
- Kosten für Prüforganisationen: Jede Prüfung durch die benannte Stelle muss bezahlt werden, wie oben bereits beschrieben. Je häufiger eine Prüfung stattfindet, desto höher werden natürlich auch die Kosten.
- Personalkosten: Wie oben beschrieben, binden Prüfungen personelle Ressourcen in der Vorbereitung, Durchführung und Nachbereitung. Vor allem, wenn Abweichungen behoben werden müssen, steigen die Kosten für Sie. Sie müssen sowohl die Zeit für die Durchführung als auch entsprechend Zeit für die Vor- und Nachbearbeitung der Audits einplanen.

Medizinprodukte: Prüfungen und Audits durch die benannte Stelle
3.2.3. Unterschied: Meldepflichten nach Markteintritt
Anforderungen an Risikoklasse I
Die Meldepflichten für Hersteller von Risikoklasse I Medizinprodukten beschränken sich im Kern auf die Anmeldung des Medizinprodukts sowie die Meldung schwerwiegender Vorkommnisse und Trends.
Zusatzanforderungen an Risikoklasse IIa
Über die Anforderungen für Risikoklasse I hinaus sind Hersteller von Produkten der Risikoklasse IIa dazu verpflichtet, einen sogenannten Periodic Safety Update Report (PSUR) zu verfassen und selbstständig an die benannte Stelle zu übermitteln. Es bietet sich an, diesen PSUR im Zuge der Post-Market-Surveillance (PMS) und Post-Market Clinical Follow-up (PMCF) zu erstellen. Auch bei Klasse I sind Sie zu PMS- und PMCF-Aktivitäten verpflichtet, allerdings entfällt in der Regel jegliche Meldepflicht.
Dieser PSUR ist für Klasse IIa Produkte mindestens alle 2 Jahre zu aktualisieren – bei höherklassigen Produkten sogar jährlich.
Auswirkungen der Risikoklasse IIa: Zeitplan
- Der Zeitplan für die Produktentwicklung wird durch die Erstellung des PSUR nicht beeinflusst.
Auswirkungen der Risikoklasse IIa: Kosten
- Personelle Ressourcen: Die Erstellung des PSUR bindet ebenfalls wieder personelle Ressourcen. Die Aufwände hängen vor allem von den zu analysierenden Daten, und etwaigen neuen Erkenntnissen ab.
4. Unternehmerische Risiken: Risikoklasse I im Vergleich zur Risikoklasse IIa
Sie wissen nun, dass eine Medizinprodukt-Zulassung unter Risikoklasse IIa erhebliche Zusatzkosten und eine starke Verzögerung bis zum Markteintritt mit sich bringt.
Wir haben aber auch schon erwähnt, dass die Bestimmung der Risikoklasse unter MDR für Software-Produkte nicht eindeutig ist und es (leider) Interpretationsspielraum gibt. Deswegen haben Hersteller von Produkten der Risikoklasse I wenig Gewissheit, dass diese Klasse auch korrekt bestimmt wurde.
Sollten Sie Ihr Produkt in die Risikoklasse I einordnen, entstehen einige Risiken aufgrund der vorhandenen Ungewissheit:
- Risiko: Aufsichtsbehörde widerspricht der Klassifizierung
- Risiko: Gerichtsverfahren nach Klage von Mitbewerbern oder Nutzern
- Risiko: Änderungen von Regularien und deren Interpretation
Auf jede dieser Risiken gehen wir in den folgenden Abschnitten kurz ein.
4.1. Risiko: Aufsichtsbehörde widerspricht der Klassifizierung
Als Hersteller eines Risikoklasse I-Produkts ist Ihre zuständige Aufsichtsbehörde dazu befugt, Sie in unregelmäßigen Abständen zu überwachen. Sie werden unangekündigten Prüfungen unterzogen.
Dabei besteht auch das Risiko, dass die Aufsichtsbehörde Ihrer Risikoklassifizierung widerspricht. Ein mögliches Szenario wäre dann Folgendes:
- Aufsichtsbehörde fragt Produktinformationen und Dokumente bei Ihnen an
- Aufsichtsbehörde prüft alle Unterlagen und kommt zum Schluss, dass das Produkt eigentlich in Risikoklasse IIa fällt
- Aufsichtsbehörde konfrontiert Sie mit der Einschätzung
- Sie diskutieren und argumentieren dagegen
- Aufsichtsbehörde lässt sich nicht überzeugen
- Aufsichtsbehörde stellt Sie vor die Wahl, die Risikoklasse zu erhöhen, oder das Produkt vom Markt zu nehmen
In diesem Fall können Sie die nachträgliche Klassifizierung nach Risikoklasse IIa angehen und sich eine benannte Stelle suchen. Dennoch sollten Sie auf diesen Fall gedanklich vorbereitet sein.
4.2. Risiko: Gerichtsverfahren nach Klage
Nicht nur Aufsichtsbehörden, sondern auch Gerichte können Einfluss auf die Risikoklasse Ihrer Produkte nehmen.
Diese sind erst dann involviert, wenn es zu einem Gerichtsverfahren kommt. Sowohl Wettbewerber als auch Nutzer Ihrer Produkte könnten potenziell in Rechtsstreit mit Ihnen treten. Dann müssen Sie sich ggf. bezüglich Ihrer Risikoklassifizierung rechtfertigen.
Szenario einer Klage eines Wettbewerbers:
Stellen Sie sich vor, Sie haben ein Software-Medizinprodukt der Risikoklasse IIa entwickelt. Nach zwei Jahren, einer klinischen Prüfung und der finalen Freigabe der benannten Stelle haben Sie es endlich auf den Markt geschafft und können nun hoffentlich mit den ersten Umsätzen die Investitionen der letzten Jahre wettmachen.
Am nächsten Tag kommt ein Mitbewerber mit einem Produkt auf den Markt, das sich von Ihrem kaum unterscheidet – außer, dass er es als Klasse I Produkt selbst zertifiziert hat. Der Mitbewerber auf das Audit einer benannten Stelle somit verzichtet und kann nun all seine Ressourcen in Marketing und Vertrieb stecken.
Was löst das bei Ihnen aus? Die natürliche Reaktion wäre ein Gefühl von Ungerechtigkeit. Und dieses Gefühl mündet manchmal in einem Gerichtsverfahren.
Fälle wie der von Dermanostic zeigen, dass die Entscheidung von Gerichten auch dazu führen können, dass Hersteller (z.B. von Klasse I Produkten) ihre Produkte vom Markt nehmen und/oder eine Hochklassifizierung durchführen müssen. Auch wenn solche Fälle eher selten sind, sollten sie als potenzielles Geschäftsrisiko berücksichtigt werden.
4.3. Risiko: Änderungen von Regularien und deren Interpretation
Die aktuelle Fassung der MDR, sowie die MDCG Guidance Dokumente (besonders das MDCG 2019-11) werden meist als Grundlage zur Bestimmung der Risikoklasse herangezogen. Doch auch die individuellen Erfahrungen von Herstellern sind eine wertvolle Quelle für die korrekte Klassifizierung.
Da aber die meisten dieser Quellen keine rechtliche Gültigkeit haben und keine Vorgabe in Stein gemeißelt ist, gibt es immer eine Chance, dass sich die Spielregeln ändern. Beispielsweise werden die folgenden Themen in der Industrie aktuell stark diskutiert:
- Eine neue Version der MDCG 2019-11 ist in Arbeit: Dieses Dokument unterstützt insbesondere Software-Hersteller bei der Bestimmung der Risikoklasse Ihres Produkts. Es enthält weitaus konkretere Informationen als die MDR und gilt somit als eine der wichtigsten Grundlagen. Auch wenn MDCG Guidance Dokumente keine rechtliche Gültigkeit haben, haben Sie dennoch sehr starken Einfluss auf die Meinungen von Prüfstellen, da diese z.T. sogar in deren Erstellung eingebunden sind.
- Etwaige Anpassungen der MDR: Aktuell gibt es zwar keine konkreten Informationen dazu, allerdings gibt es immer wieder Bestrebungen, Teile der MDR zu ändern. Darunter könnten auch die Klassifizierungsregeln fallen, welche von Herstellern immer wieder beklagt werden.
- Höhere Instanzen nehmen Einfluss auf die einzelnen Landesaufsichtsbehörden: Das BfArM oder sogar das Bundesgesundheitsministerium (BGM) könnten Einfluss auf Ihre Aufsichtsbehörde nehmen. Eine strengere Auslegung der Regel 11 von höheren Instanzen kann also durchaus zu einer erhöhten Aufmerksamkeit oder sogar verschärfenden Maßnahmen Ihrer zuständigen Aufsichtsbehörde führen. Darunter kann auch die höhere Klassifizierung Ihrer Produkte zählen.
5. Fazit
Die Kosten und die Zeit bis zum Markteintritt eines neuen Produkts unterscheiden sich zwischen Risikoklasse I und Risikoklasse IIa sehr stark.
- Zeitrahmen bis Markteintritt: Die Zulassung einer Software der Klasse IIa kann mehr als ein Jahr länger dauern als bei einem Medizinprodukt der Klasse I.
- Kosten: Durch die Kosten für die benannte Stelle und die Bindung interner Ressourcen muss mit Zusatzkosten von 100.000 € und mehr bei Risikoklasse IIa gerechnet werden. Hinzu kommen Opportunitätskosten für ausbleibende Umsätze durch die Verzögerung beim Markteintritt.
Was für Großkonzerne erstmal kein Problem darstellt, wird hier für Start-ups zu einer existenziellen Bedrohung. Die Anforderungen der Risikoklasse IIa sind für viele junge Unternehmen nicht stemmbar und führen zur Insolvenz. Das führt dazu, dass viele innovative Produkte nie auf den Markt kommen und die Versorgung verbessern können.
Es ist also für Deutschland und Europa enorm wichtig, dass auch in Zukunft für Software-Produkte mit niedrigen Risiken die Kategorisierung in Risikoklasse I möglich ist. Die Risiken, die z.B. von digitalen Gesundheitsanwendungen (DiGA) ausgehen, sind in der Regel sehr gering. Diese Anwendungen haben aber einen enormen positiven Einfluss auf die Patientenversorgung. Wir hoffen daher, dass dies bei der zukünftigen Aktualisierung der MDCG-Klassifizierungs-Guideline bedacht wird.
In der Zwischenzeit soll Ihnen die obige Guideline dabei helfen, die aktuelle Situation in Bezug auf die Klassifizierung von Software-Medizinprodukten besser zu verstehen.
Wenn Sie eine medizinische Software planen und einen Entwicklungspartner suchen, kontaktieren Sie uns gerne. Als spezialisierter Dienstleister liegt unser Fokus auf der regulatorisch-konformen Entwicklung von medizinischen Apps und Gesundheitssoftware. Wir helfen Ihnen, ein Produkt umzusetzen, das am Ende auch erfolgreich auf dem Markt landet und Nutzern nachhaltig hilft.
The post MDR Risikoklasse I vs. IIa: Unterschiede für Software-Medizinprodukte appeared first on QuickBird Medical.
]]>The post Software-Medizinprodukt ohne Qualitätsmanagement & Regulatorik zulassen? appeared first on QuickBird Medical.
]]>Für viele Unternehmen stellt sich daher die Frage:
Gibt es einen Weg, die regulatorischen Pflichten für Medizinprodukt-Hersteller komplett an einen Dienstleister auszulagern?
Ja, den gibt es: Die Auslagerung der sogenannten Legalhersteller-Rolle an ein externes Unternehmen. Dies ist eine effiziente und risikominimierende Lösung, um Ihre Ressourcen zu schonen und gleichzeitig regulatorische Sicherheit zu schaffen.
In diesem Leitfaden …
- … beleuchten wir die Vorteile eines externen Legalherstellers für Ihr Produkt,
- … beantworten wir die wichtigsten Fragen rund um diesen Ansatz und
- … sehen uns an, für welche Unternehmen dieses Modell wirklich Sinn ergibt.
Inhaltsverzeichnis
- 1. Problem: Pflichten für Legalhersteller von Medizinprodukten
- 2. Lösung: Externer Inverkehrbringer für MDR-Medizinprodukte
- 3. Vorteile eines externen Herstellers nach MDR
- 4. Ablauf einer solchen Zusammenarbeit zur Auslagerung der Legalhersteller-Rolle
- 5. Wichtige Fragen und Antworten
- 5.1. Ich möchte die Hersteller-Rolle auslagern, um das Produkt schneller auf den Markt zu bringen. Später möchte ich die Hersteller-Rolle für das Produkt aber wieder selbst übernehmen. Ist das möglich?
- 5.2. Behalte ich alle Besitzrechte am Produkt?
- 5.3. Ist das teurer als die Themen inhouse zu übernehmen?
- 5.4. Wer haftet für das Produkt im regulatorischen Sinne, z.B. bei Patientenschäden?
- 5.5. Kann die Hersteller-Rolle für eine DiGA ausgelagert werden?
- 5.6. Kann ich meine Hersteller-Rolle für ein zertifiziertes Medizinprodukt nachträglich auslagern?
- 5.7. Kann ich das Produkt nach Inverkehrbringen weiterentwickeln?
- 5.8. Kann ich weiterhin ein eigenes Entwicklerteam nutzen?
- 6. Beispiele für Medizinprodukte-Hersteller mit externem Inverkehrbringer
- 7. Fazit: Für wen lohnt es sich, die Hersteller-Rolle auszulagern?
1. Problem: Pflichten für Legalhersteller von Medizinprodukten
Wenn Sie eine Software entwickeln möchten, die als Medizinprodukt zertifiziert werden soll, kommen erhebliche Aufwände auf Sie zu. Der Hersteller eines Software-Medizinprodukts muss nach MDR u.a. folgende Pflichten erfüllen:
- Aufbau eines Qualitätsmanagementsystems (QMS): Aufbau der Prozesse und Strukturen (nach MDR & ISO 13485)
- Erstellung der technischen Dokumentation: Einhaltung der MDR, IEC 62304, IEC 62366-1, ISO 13485, IEC 82304, ISO 14971 (um hier nur die wichtigsten Normen und Gesetze zu nennen)
- Haftung für das Produktrisiko im Rahmen der MDR: Sie haften als Hersteller bei Patientenschäden oder Konformitätsverletzungen Ihrer regulatorischen Pflichten. Bei fahrlässigem Handel haften Sie ggf. sogar persönlich im Sinne des Strafrechts.
- Kontinuierliche Durchführung aller Qualitätsmanagementprozesse: Post-Market-Surveillance (PMS), Post-Market-Clinical-Follow-Up (PMCF), Vigilanz, Management Reviews, interne Audits, Schulungen, Computerized Systems Validation (CSV), Risikomanagement, klinische Bewertung usw.
- Überwachung der Informationssicherheit: Sie müssen die Informationssicherheit Ihres Produkts und Ihrer Firma durch strukturierte Prozesse und Maßnahmen im Griff haben.
Wenn Sie noch nie ein Medizinprodukt nach MDR in Verkehr gebracht haben, kommen damit erhebliche Aufwände und Pflichten auf Sie zu.
2. Lösung: Externer Inverkehrbringer für MDR-Medizinprodukte
Wenn Sie die oben genannten Pflichten nicht erfüllen möchten oder können, gibt es eine sinnvolle Alternative: die Rolle des Herstellers nach MDR an einen Dienstleister auszulagern.
So können Sie sich auf Ihre Kernkompetenzen konzentrieren, während der Dienstleister die volle Verantwortung für das regulatorische Inverkehrbringen Ihres Software-Medizinprodukts im Rahmen der MDR übernimmt.
QuickBird Medical als externer Inverkehrbringer für Ihr Software-Produkt
Hinweis: Im Rahmen dieses Artikels werden die Begriffe „Legalhersteller“, „Hersteller“ und „Inverkehrbringer“ synonym verwendet.
2.1. Händler und Hersteller für Ihr Software-Medizinprodukt
Je nach Verantwortlichkeitsverteilung ergeben sich unterschiedliche regulatorische Rollen. Eine übliche Rollenverteilung ist es, dass wir bei QuickBird Medical die Rolle des sogenannten Legalherstellers nach MDR übernehmen und Sie die Rolle des sogenannten Händlers übernehmen.
Dementsprechend haftet QuickBird Medical für alle Pflichten, die sich nach MDR auf den Medizinprodukt-Hersteller beziehen.
Sie können dann ihr eigenes Logo/Design/Branding auf dem Produkt platzieren, aber die regulatorischen Aufwände erfolgreich auslagern.
Als Händler übernehmen Sie ggf. dann den Kunden-Support, Vertrieb und Marketing des Produkts. Auch hier gibt es z.B. im Rahmen des Heilmittelwerbegesetzes einige Pflichten, die Sie beachten müssen. Wir schulen Sie in diesen Themen und unterstützen Sie. So reduziert sich Ihr Aufwand in Bezug auf regulatorische Themen auf das absolute Minimum.
2.2. OEM und PLM für Ihr Software-Medizinprodukt
Im Rahmen der früher geltenden Medical Device Directive (MDD) war die sogenannte OEM-PLM-Konstellation gängig:
- OEM (Original Equipment Manufacturer): Der OEM ist der tatsächliche Entwickler und Produzent des Software-Medizinprodukts. Er erstellt die vollständige technische Dokumentation, führt alle notwendigen Konformitätsbewertungsverfahren durch und bringt das Produkt zunächst gesetzeskonform zur Marktreife. Der OEM trägt somit die regulatorische Verantwortung für die Produktsicherheit und -konformität.
- PLM (Private Label Manufacturer): Der PLM übernimmt das fertige Medizinprodukt vom OEM und bringt es unter eigenem Namen, Logo und Branding auf den Markt.
Nach Ablösung der MDD durch die MDR gibt es die OEM-PLM-Konstellation in ihrer ursprünglichen Form nicht mehr. Stattdessen spricht man von einer Händler-Hersteller-Konstellation als Alternative, welche in den vorangegangenen Kapiteln beschrieben wird. So lassen sich dieselben Ziele weiterhin auch unter MDR erreichen.
3. Vorteile eines externen Herstellers nach MDR
Die Auslagerung der Hersteller-Rolle an einen externen Legalhersteller bietet u.a. folgende Vorteile:
- Schneller und einfacher Marktzugang: Ein externer Hersteller ermöglicht es, Produkte deutlich schneller auf den Markt zu bringen, da er über bestehende Zertifizierungen (z. B. ISO 13485) und erprobte Qualitätsmanagementprozesse verfügt. Dies spart Zeit und vermeidet Verzögerungen durch aufwendige interne Vorbereitungen.
- Entlastung von Ressourcen und Kosten: Die regulatorischen Pflichten werden vollständig ausgelagert, wodurch Unternehmen interne Ressourcen wie Personal und finanzielle Mittel für andere Projekte freisetzen können. Die hohen Kosten für die Einrichtung und Pflege eines QMS, Zertifizierungen sowie Audits entfallen oder werden erheblich reduziert.
- Regulatorische Sicherheit und Risikominimierung: Der externe Hersteller bringt umfassende regulatorische Expertise mit und sorgt dafür, dass Produkte den neuesten regulatorischen Anforderungen entsprechen. Auch das Risiko von Haftung bei Produktrückrufen oder Sicherheitsvorfällen wird ausgelagert. Zudem bleibt der externe Hersteller für Anpassungen an neue Regularien verantwortlich, was die Unsicherheit für das Unternehmen minimiert.
- Flexibilität und Fokus auf Kernkompetenzen: Unternehmen können sich auf ihre Stärken wie Produktentwicklung und Innovation konzentrieren, ohne durch regulatorische Anforderungen abgelenkt zu werden. Gleichzeitig bietet ein externer Hersteller Flexibilität in der Organisationsstruktur, indem er die regulatorischen Pflichten übernimmt.
- Strategische Vorteile: Durch die Zusammenarbeit mit einem externen Hersteller erhalten Unternehmen Zugang zu branchenspezifischen Netzwerken und erprobten Prozessen, die den Markteintritt erleichtern. Außerdem entfällt die Notwendigkeit, regulatorisches Wissen intern aufzubauen und zu pflegen, was den Aufwand erheblich reduziert.
Diese Vorteile machen die Auslagerung der Hersteller-Rolle zu einer attraktiven Option, insbesondere für Unternehmen, die schnelle Ergebnisse erzielen und gleichzeitig Risiken sowie Komplexität minimieren möchten.
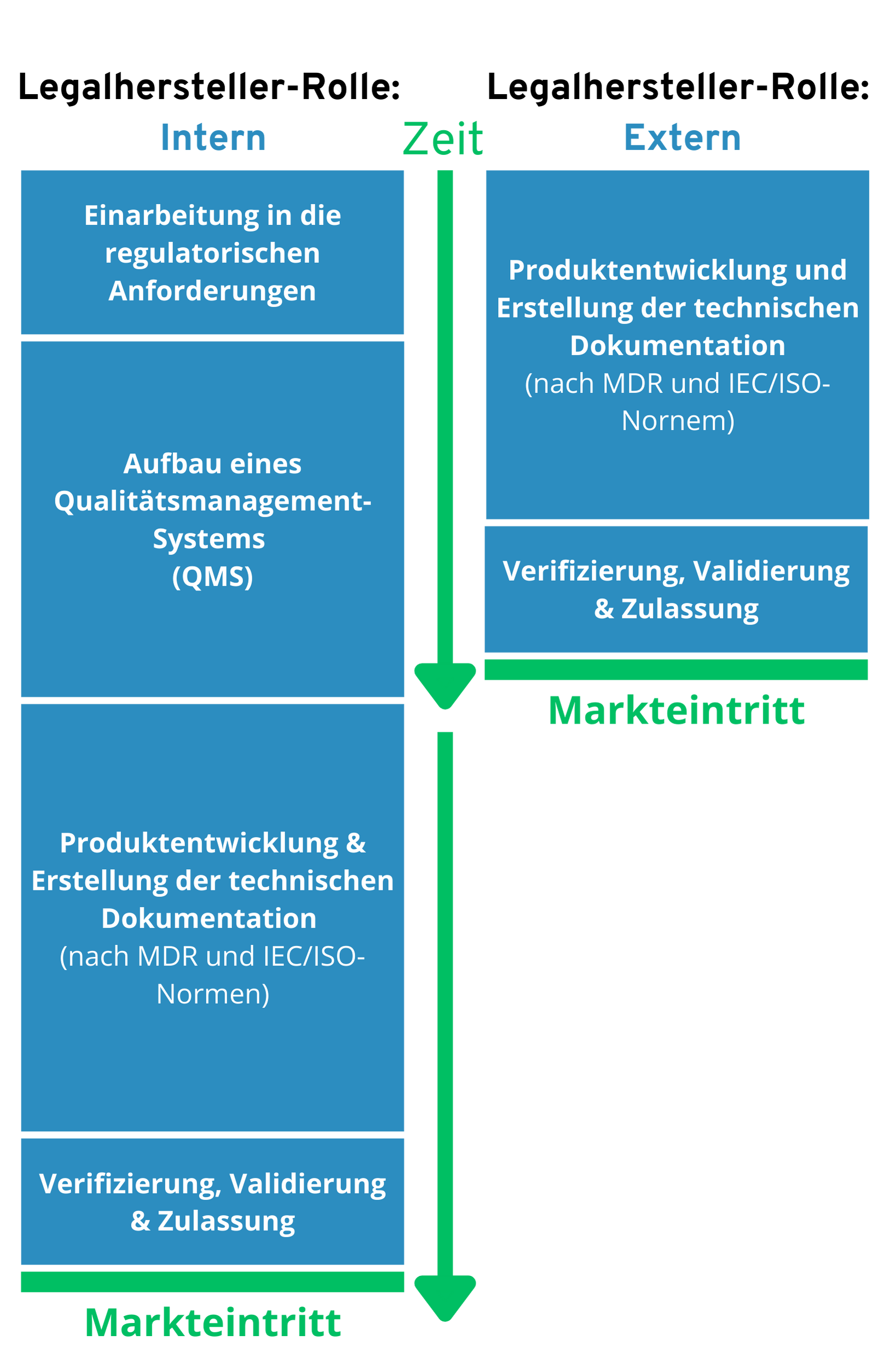
Zeit bis Markteintritt – Vergleich:
Legalhersteller-Rolle Intern vs. Extern
(Beispielhaft für Risikoklasse I nach MDR)
4. Ablauf einer solchen Zusammenarbeit zur Auslagerung der Legalhersteller-Rolle
Wenn Sie die Auslagerung der Hersteller-Rolle mit uns als Dienstleister angehen möchten, erstellen wir zu Beginn einen individuellen Projektplan, der auf Ihre Situation zugeschnitten ist. Wichtig ist initial erstmal die Bestimmung der Verantwortlichkeitsverteilung. Hierfür sollten u.a. folgende Fragen beantworten:
- Möchten Sie die Software-Entwicklung auslagern oder Ihr eigenes, internes Team hierfür nutzen? Beide Optionen sind grundsätzlich möglich. Wir können die gesamte Produktentwicklung von technischer und regulatorischer Seite für Sie übernehmen oder auch nur einzelne Leistungen ergänzen, die Sie nicht intern abbilden können.
- Möchten Sie die Content-Entwicklung für die Software (medizinische Texte, Bilder, Videos, Audio) selbst übernehmen oder auslagern? Beide Optionen sind prinzipiell möglich. Es ist auch möglich, dass Sie den Content inhaltlich vorbereiten und wir die finale Editierung und visuelle Gestaltung übernehmen.
- Möchten Sie den Kunden-Support (Kontakt-Hotline) selbst anbieten oder auch an uns als Dienstleister auslagern? Beide Optionen sind prinzipiell möglich. Wenn Sie diesen Service inhouse übernehmen, schulen wir Sie in Bezug auf die relevanten regulatorischen Pflichten.
Nach Bestimmung der Verantwortlichkeitsverteilung besteht der finale Projektplan z.B. aus den folgenden Schritten:
- 1. Definition der Zweckbestimmung und Risikoklasse nach MDR
- 2. Produktentwicklung:
- a. Erstellung der technischen Dokumentation unter Einhaltung aller Normen & Standards
- b. Software-Entwicklung unter Einhaltung aller Normen & Standards
- 3. Verifizierung & Validierung des Produkts (inkl. klinische Bewertung, summative Usability-Evaluation, Cyber-Security-Validierung etc.)
- 4. Registrierung des Medizinprodukts durch Selbstzertifizierung (Risikoklasse I) bzw. Konformitätsbewertungsverfahren mit benannter Stelle (Risikoklasse IIa und höher)
- 5. Weiterentwicklung des Produkts sowie Überwachung nach dem Inverkehrbringen (Post Market Surveillance, Post-Market-Clinical-Follow-Up, Vigilanz, Wartung, Betrieb, …)
Achtung: Dieser Ablauf ist stark vereinfacht und soll Ihnen nur ein Grundverständnis geben.
Für alle genannten Schritte übernehmen wir gern die federführende Rolle und leiten Sie durch den Prozess. Selbstverständlich haben Sie weiterhin die volle Kontrolle und treffen die relevanten Geschäftsentscheidungen.
Kontakt aufnehmen:
Sie möchten die Legalhersteller-Rolle auslagern?
Wir bieten diesen Service für Software-Medizinprodukte und DiGA an.
Kontaktieren Sie uns hierzu: Mehr Informationen zur Kontaktaufnahme
5. Wichtige Fragen und Antworten
Hier gehen wir auf wichtige Fragen ein, die bei der Auslagerung der Hersteller-Rolle erfahrungsgemäß aufkommen.
5.1. Ich möchte die Hersteller-Rolle auslagern, um das Produkt schneller auf den Markt zu bringen. Später möchte ich die Hersteller-Rolle für das Produkt aber wieder selbst übernehmen. Ist das möglich?
Ja, das ist absolut möglich. Sie behalten die Intellectual Property (IP) am Produkt und der gesamten Dokumentation. Daher ist es für Sie zu jedem Zeitpunkt möglich, das Produkt umzumelden und selbst die Hersteller-Rolle zu übernehmen. So profitieren Sie von einem schnellen Marktstart Ihres Produkts und können die Regulatorik mittelfristig wieder inhouse übernehmen. Wir helfen Ihnen dabei, diesen Übergang reibungslos zu absolvieren.
5.2. Behalte ich alle Besitzrechte am Produkt?
Ja, sie haben die komplette Intellectual Property (IP) am Produkt und der Dokumentation. QuickBird Medical bietet Ihnen lediglich einen Service an, um Ihnen beim Launch eines regulierten Software-Medizinprodukts zu helfen. Sie haben die volle Kontrolle und Besitzrechte am Produkt.
5.3. Ist das teurer als die Themen inhouse zu übernehmen?
Das hängt natürlich von einer Vielzahl von Faktoren ab. Allerdings ist es unsere Erfahrung, dass die Auslagerung der Hersteller-Rolle insgesamt zu Kosteneinsparungen führt.
Aus folgenden Gründen können Kosteneinsparungen realisiert werden:
- Kein Aufbau eines QMS nötig: Das Qualitätsmanagementsystem (QMS) existiert bereits und ist bei QuickBird Medical nach ISO 13485 zertifiziert. Sie müssen keine Kosten für den Aufbau und die Zertifizierung eines QMS zahlen.
- Kein Aufbau eines ISMS nötig: Das Informationssicherheitsmanagementsystem (ISMS) existiert bereits und ist bei QuickBird Medical nach ISO 27001 zertifiziert. Sie müssen keine Kosten für den Aufbau und die Zertifizierung eines ISMS zahlen.
- Geschultes, effizientes Team: Das QuickBird Medical-Team ist durch zahlreiche Projekte sehr effizient in der Erstellung der technischen Dokumentation für Software-Medizinprodukte. Daher fallen geringere Aufwände an, als wenn ein ungeschultes Team diese Aufgabe übernehmen würde.
- Auslastungsspitzen und -täler: Nach Veröffentlichung des Produkts wird der Aufwand für das Regulatorik- und Software-Team erfahrungsgemäß geringer als für den initialen Launch. Für das externe Team fallen dann nur Kosten an, wenn auch Arbeit getan wird. Eine interne Vollzeit-Person muss kontinuierlich bezahlt werden – auch wenn die Arbeitslast niedriger wird.
5.4. Wer haftet für das Produkt im regulatorischen Sinne, z.B. bei Patientenschäden?
Im Rahmen der regulatorischen Pflichten nach MDR übernimmt der externe Inverkehrbringer (z. B. QuickBird Medical) die volle Haftung für das Produkt. Das bedeutet, QuickBird Medical gewährleistet nicht nur die Einhaltung aller regulatorischen Anforderungen, sondern trägt auch die Verantwortung im Schadensfall – etwa bei Patientenschäden. Sie können das mit Medizinprodukten verbundene Risiko also auf diese Weise erfolgreich auslagern.
QuickBird Medical besitzt als Medizinprodukt-Hersteller eine rechtlich verbindliche Haftpflichtversicherung für Personenschäden. Außerdem werden wir 4-mal jährlich durch externe Parteien auditiert, um die regulatorische Compliance sicherzustellen.
5.5. Kann die Hersteller-Rolle für eine DiGA ausgelagert werden?
Ja, QuickBird Medical ist sowohl nach ISO 13485 für Qualitätsmanagement als auch nach ISO 27001 für Informationssicherheitsmanagment zertifiziert. Damit erfüllen wir die Voraussetzungen, um als DiGA-Hersteller aufzutreten und übernehmen diesen Service auch schon für existierende DiGA-Kunden.
5.6. Kann ich meine Hersteller-Rolle für ein zertifiziertes Medizinprodukt nachträglich auslagern?
Ja, das ist möglich. Wir überprüfen und überführen dann Ihre existierende technische Dokumentation. Danach kann das Medizinprodukt zu uns als neuen Legalhersteller umgemeldet werden. Je nachdem, ob eine benannte Stelle involviert ist, müssen weitere Formalitäten beachtet werden.
5.7. Kann ich das Produkt nach dem Inverkehrbringen weiterentwickeln?
Sie können natürlich jederzeit Änderungen am Produkt vornehmen. Diese Erweiterungen werden bewertet und durchlaufen dann unsere Qualitätsmanagementprozesse vor der Veröffentlichung, um die regulatorische Compliance sicherzustellen. Das Ganze funktioniert in einem agilen Entwicklungszyklus zur regelmäßigen Veröffentlichung neuer Funktionen.
5.8. Kann ich weiterhin ein eigenes Entwicklerteam nutzen?
QuickBird Medical kann für Sie die gesamte Entwicklung sowie die externe Legalhersteller-Rolle nach MDR übernehmen.
Auf der anderen Seite ist es aber auch möglich, ein internes Entwicklerteam bei Ihnen für die Umsetzung des Produkts zu nutzen und weiterhin die Hersteller-Rolle auszulagern.
6. Beispiele für Medizinprodukte-Hersteller mit externem Inverkehrbringer
Um das Ganze ein wenig greifbarer zu machen, finden Sie hier zwei Beispiele für Firmen, für welche QuickBird Medical die Legalhersteller-Rolle übernimmt:
- Mamly: Mamly ist eine zugelassene Medizinprodukt-App zur Behandlung von peripartaler Depression. Sie hilft Schwangeren mit Achtsamkeitstraining und professioneller Unterstützung dabei, psychische Belastungen zu reduzieren.
Zur Case-Study - Kontina: Kontina ist eine zugelassene Medizinprodukt-App zur Behandlung einer überaktiven Blase (OAB) durch Training, Lerninhalte und ein Blasentagebuch zur Symptomreduktion.
Zur Case-Study
7. Fazit: Für wen lohnt es sich, die Hersteller-Rolle auszulagern?
Wann lohnt es sich gegebenenfalls nicht, die Rolle des Legalherstellers auszulagern?
- Sie haben bereits viel Regulatorik-Expertise inhouse und ein Qualitätsmanagement-Team, das sich mit Software-Medizinprodukten auskennt? -> Dann sollten Sie dieses Team nutzen und selbst der Legalhersteller werden.
- Sie haben mehrere Software-Medizinprodukte auf dem Markt -> Je mehr Software-Medizinprodukte Sie haben, desto höher werden die Synergie-Effekte eines eigenen Qualitätsmanagement-Teams.
Wann lohnt es sich? Wenn Sie folgende Ziele haben, kann sich die Auslagerung der Legalhersteller-Rolle an uns als Dienstleister lohnen:
- Sie möchten möglichst schnell Ihr Produkt auf den Markt bringen, um Umsätze zu generieren.
- Sie möchten regulatorische Komplexität und interne Aufwände vermeiden, die eine Distraktion vom Kerngeschäft darstellen.
- Sie möchten rechtliche Haftungsrisiken für das Medizinprodukt auslagern.
Wichtig ist, dass Sie sich strategisch überlegen, welches Szenario für Sie mehr Sinn ergibt.
Gerne beraten wir Sie dabei. Wenn Sie eine medizinische Software planen und einen Partner für die Umsetzung suchen, kontaktieren Sie uns jederzeit. Als spezialisierter Dienstleister liegt unser Fokus auf der regulatorisch-konformen Entwicklung von medizinischen Apps und Gesundheitssoftware.
Kontakt aufnehmen:
Sie möchten die Legalhersteller-Rolle auslagern?
Wir bieten diesen Service für Software-Medizinprodukte und DiGA an.
Kontaktieren Sie uns hierzu: Mehr Informationen zur Kontaktaufnahme
The post Software-Medizinprodukt ohne Qualitätsmanagement & Regulatorik zulassen? appeared first on QuickBird Medical.
]]>The post Erstattung von Software im GKV-Hilfsmittelverzeichnis (HMV) appeared first on QuickBird Medical.
]]>Die Realität für viele Digital Health-Unternehmen: Ohne Erstattung durch die gesetzlichen Krankenkassen lässt sich im deutschen Gesundheitssystem schwer genug Geld verdienen, um langfristig als Firma zu überleben. Die Frage, die sich viele Hersteller stellen, lautet deshalb: Gibt es neben den bekannten Modellen weitere Wege, die Tür zur Erstattung zu öffnen?
Ein Ansatz, der bislang nur wenig Beachtung findet, ist die Listung von Software im Hilfsmittelverzeichnis (HMV) der GKV. Ursprünglich für physische Produkte wie Rollatoren, Hörgeräte oder Prothesen gedacht, stellt sich die spannende Frage, ob auch digitale Lösungen diesen Weg gehen können – und ob sich der Aufwand lohnt, ein Softwareprodukt als „Hilfsmittel“ zu positionieren.
In diesem Artikel zeigen wir, warum das Hilfsmittelverzeichnis ein potenzieller Weg zur Erstattung für Softwareprodukte ist, welche Anforderungen digitale Anwendungen erfüllen müssen, wie der Aufnahmeprozess abläuft und ob sich der Aufwand für Hersteller lohnt.
Inhaltsverzeichnis
- 1. Vorteile für Hersteller bei Listung im Hilfsmittelverzeichnis
- 2. Definition eines Hilfsmittels
- 3. Das GKV-Hilfsmittelverzeichnis
- 4. Schritte zur Zulassung im Hilfsmittelverzeichnis
- 5. Qualifizierung: Eignung ihrer Software für das Hilfsmittelverzeichnis
- 6. Anforderungen an Software im Hilfsmittelverzeichnis
- 7. Antrag zur Aufnahme ins Hilfsmittelverzeichnis
- 8. Vergütung und Preisfindung mit den Krankenkassen
- 9. Fortschreibung der Produktgruppen
- 10. Beispiele: Gelistete Software-Produkten im Hilfsmittelverzeichnis
- 11. Warum gibt es kaum Standalone-Software im Hilfsmittelverzeichnis? (Hürden)
- 12. Weitere Wege in die Kostenerstattung durch Krankenkassen
- 13. Fazit: Lohnt sich das Hilfsmittelverzeichnis für Software-Hersteller?
1. Vorteile für Hersteller bei Listung im Hilfsmittelverzeichnis
Die Listung eines Produkts im Hilfsmittelverzeichnis der GKV bietet Ihnen als Hersteller einen zentralen Vorteil gegenüber z. B. Selektivverträgen: Alle gesetzlichen Krankenkassen in Deutschland sind verpflichtet, Ihr Produkt zu erstatten. Sie müssen daher nicht separat jede Krankenkasse von Ihrem Produkt überzeugen, sondern können einem zentralisierten Antragsprozess folgen.
Bevor die tatsächliche Kostenerstattung erfolgt, müssen Hersteller jedoch die Vergütungshöhe klären. Hier gibt es zwei Optionen: Entweder tritt der Hersteller einem bestehenden Vertrag für eine Produktgruppe bei – dies garantiert einen schnellen Marktzugang, kann aber festgelegte Vergütungssätze bedeuten, die nicht immer ideal für Softwareprodukte sind. Alternativ können individuelle Vereinbarungen mit jeder Krankenkasse getroffen werden, was jedoch zeit- und ressourcenintensiv ist.
Dies ist ein entscheidender Unterschied zum DiGA-Verfahren, in welchem die Höhe der Erstattungsbeträge einmalig mit dem GKV-Spitzenverband (GKV-SV) verhandelt wird und dann für die Erstattung durch alle Krankenkassen gilt.
| Aspekt | Hilfsmittelverzeichnis | DiGA | Selektivverträge |
| Garantierter Zugang zu allen Krankenkassen | Ja, Antrag erlaubt Erstattung durch alle Krankenkassen | Ja, Antrag erlaubt Erstattung durch alle Krankenkassen | Nein, Selektivvertrag muss einzeln mit jeder Krankenkasse verhandelt werden |
| Zentralisierte Vergütungsverhandlung | Nein, Vergütung individuell mit jeder Kasse vereinbaren oder existierenden Verträgen beitreten | Ja, Vergütungshöhe zentral mit GKV-SV verhandelt und einheitlich | Nein, Vergütung individuell mit jeder Kasse vereinbaren |
Trotzdem bleibt die Listung im Hilfsmittelverzeichnis eine attraktive Möglichkeit, digitale Produkte flächendeckend für Versicherte verfügbar zu machen. Die setzt jedoch voraus, dass Ihr Produkt in die Definition eines Hilfsmittels fällt.
2. Definition eines Hilfsmittels
Laut Definition des Gemeinsamen Bundesausschusses (G-BA) sind Hilfsmittel Gegenstände, die „im Einzelfall erforderlich sind, um durch ersetzende, unterstützende oder entlastende Wirkung den Erfolg einer Krankenbehandlung zu sichern, einer drohenden Behinderung vorzubeugen oder eine Behinderung auszugleichen”.
Hilfsmittel können in ihrer Funktion u. a. als “Körperersatzstück” (§33 SGB V) dienen. Sie haben also die primäre Aufgabe, Menschen dabei zu unterstützen, eine spezifische Tätigkeit auszuführen, eine Einschränkung zu kompensieren oder ein Ziel zu erreichen, das ohne das Hilfsmittel schwieriger oder unmöglich wäre.
Allgemeine Gebrauchsgegenstände des täglichen Lebens – bspw. ein Fieberthermometer – können kein Hilfsmittel sein. Ebenso wenig die in §34 SGB V ausgeschlossenen Hilfsmittel.
Klassische Hilfsmittel sind beispielsweise Rollstühle, Hörgeräte, Inkontinenzhilfen, Kompressionsstrümpfe oder Prothesen. Es sind also oft physische Gegenstände. Tatsächlich können aber auch reine Software-Produkte oder Apps in die Definition eines Hilfsmittels fallen. Das bestätigte das Bundessozialgericht erstmals im Jahr 2001.
 Unterschied der Ziele zwischen DiGA und Hilfsmitteln
Unterschied der Ziele zwischen DiGA und Hilfsmitteln
Zusammenfassend haben Hilfsmittel also das Ziel,
… den Erfolg einer Krankenbehandlung zu sichern (§ 33 SGB V),
… eine drohende Behinderung vorzubeugen (§ 33 SGB V und § 47 SGB V)
… eine bereits vorhandene Behinderung auszugleichen (§ 33 SGB V und § 47 SGB V)
… eine Pflegebedürftigkeit zu vermeiden (§ 47 SGB V).
Erstattungsfähige Hilfsmittel sind im Hilfsmittelverzeichnis des GKV-Spitzenverbands gelistet.
3. Das GKV-Hilfsmittelverzeichnis
Das Hilfsmittelverzeichnis wird vom GKV-Spitzenverband gemäß § 139 SGB V erstellt und regelmäßig aktualisiert. Es listet Hilfsmittel auf, deren Kosten von der gesetzlichen Krankenversicherung (GKV) übernommen werden können.
Das Hilfsmittelverzeichnis ist vergleichbar mit einem Katalog. Es dient u. a. als Orientierungshilfe für Versicherte und Leistungserbringer, indem es über erstattungsfähige Hilfsmittel sowie deren Art und Qualität informiert.
Das GKV-Hilfsmittelverzeichnis ist wie folgt gegliedert:
Produktgruppen > Untergruppen > Anwendungsort > Produktarten > Produkte

Screenshot aus dem GKV-Hilfsmittelverzeichnis
Das Hilfsmittel-Verzeichnis (HMV) ist öffentlich zugänglich unter: https://hilfsmittel.gkv-spitzenverband.de/home
Die Nummerierung der Produkte im Hilfsmittelverzeichnis folgt einer systematischen und hierarchischen Struktur, die eine klare Zuordnung und Übersichtlichkeit gewährleistet. Die Nummer setzt sich aus mehreren Ziffern zusammen: Die ersten beiden Ziffern kennzeichnen die Hauptproduktgruppe, die nächsten beiden Stellen spezifizieren den Anwendungsort sowie die Untergruppen. Am Ende steht die vierstellige, individuelle Produktnummer, wobei die erste Ziffer hierbei nochmals die Produktart klassifiziert.

Beispiel für die Produktnummerierung im Hilfsmittelverzeichnis –
Bildquelle: https://www.rehadat-gkv.de/hinweise/aufbau-des-verzeichnisses/
4. Schritte zur Zulassung im Hilfsmittelverzeichnis

Prozess der Beantragung eines Softwareprodukts für das GKV-Hilfsmittelverzeichnis
In der obigen Abbildung ist der Prozess von Anfang bis zur erfolgreichen Vergütung skizziert. Die wichtigsten Schritte sind Folgende:
- Qualifizierung: Eignet sich mein Produktkonzept überhaupt für die Aufnahme in das Hilfsmittelverzeichnis?
- Erfüllung der Anforderungen: Sie müssen alle allgemeinen und Produkt-spezifischen Anforderungen umsetzen, die zur Aufnahme ins HMV nötig sind.
- Antragsstellung: Sie müssen nun den Antrag ausfüllen und abschicken.
- Prüfung und Entscheidung: Sie erhalten innerhalb der gesetzlichen Fristen eine Rückmeldung und Entscheidung in Bezug auf Ihren Antrag.
- Vergütung mit Krankenkasse: Nach erfolgreicher Aufnahme in das HMV müssen Sie die Höhe der Vergütung mit den verschiedenen Krankenkassen bestimmen.
Nach Schritt 5 sind Sie erstattungsfähig und können über Ihr Produkt Einnahmen generieren. Alle Schritte dieses Prozesses werden in den folgenden Kapiteln im Detail erläutert.
5. Qualifizierung: Eignung Ihrer Software für das Hilfsmittelverzeichnis
Um herauszufinden, ob Ihr Produkt in das Hilfsmittelverzeichnis aufgenommen werden kann, sollten Sie folgende Dinge prüfen.
5.1 Qualifizierungs-Check: Zweck Ihres Produkts
Wir haben zu Beginn bereits erfahren, dass auch Softwareprodukte unter die „Hilfsmittel“-Definition fallen können und dadurch auch im Hilfsmittelverzeichnis gelistet werden können.
Die große Herausforderung für Softwareprodukte besteht darin, nachzuweisen, dass die Software den Hilfsmittel-Charakter erfüllt – also bestimmten Behinderungen vorbeugt oder diese kompensiert. Wichtig ist, zu verstehen: Hilfsmittel sind keine Therapien. Sie können und sollen natürlich in ihrer Funktion, „den Erfolg einer Krankenbehandlung zu sichern“ therapiebegleitend sein, aber ihre primäre Aufgabe ist die Sofort-Hilfe.
Um herauszufinden, ob Ihr Produkt im Hilfsmittelverzeichnisses gelistet werden kann, müssen Sie zuallererst prüfen, ob es die Kriterien eines Hilfsmittels erfüllt. Die Definition eines Hilfsmittels haben wir in dem obigen Abschnitt näher erläutert.
5.2 Qualifizierungs-Check: Kategorisierung zu einer Produktgruppe
Wenn Ihr Produkt potenziell in die Definition eines Hilfsmittels fällt, kommen wir zu Schritt 2: Es gilt nun eine existierende Produktgruppe im Hilfsmittelverzeichnis zu finden, in das Ihr Produkt hineinpasst.
Vorneweg: Aktuell gibt es keine eigene Produktgruppe für Standalone-Softwareprodukte oder mobile Apps im Hilfsmittelverzeichnis.
Das bedeutet: Wenn Sie eine Software als Hilfsmittel beantragen wollen, müssen Sie sich die einzelnen Produktgruppen ansehen und die jeweils individuellen Anforderungsprofile überprüfen.
Was, wenn Sie in keine Produktgruppe oder -Art passen? Falls Sie in keine existierende Produktgruppe passen, bleibt nur noch die Möglichkeit eine neue Produktgruppe zu beantragen. Dieser Prozess ist aber enorm aufwändig und zieht sich potenziell über viele Jahre. Daher ist dieser Weg für kaum einen Software-Hersteller erstrebenswert.
Der GKV schreibt zur Berücksichtigung von Innovationen im Hilfsmittelverzeichnis:
“Es werden auch neue Produkte zur Aufnahme in das Hilfsmittelverzeichnis angemeldet, die in die bestehende Systematik nicht eingeordnet werden können, da eine entsprechende Produktuntergruppe bzw. -art noch nicht gebildet wurde. Daher wäre vor der Listung eines solchen Produktes zunächst eine Fortschreibung der betreffenden Produktgruppe erforderlich. Dies erfordert Zeit, da alle Beteiligungsrechte und Verfahrensschritte eingehalten werden müssen.”
Sie sollten also unbedingt eine existierende Produktgruppe finden. Falls dies nicht möglich ist, ist ggf. eher ein komplett anderer Erstattungsweg z.B. über Selektivverträge für Sie ratsam.
6. Anforderungen an Software im Hilfsmittelverzeichnis
Blicken wir zunächst auf die gesetzlich definierten Anforderungen. Das Sozialgesetzbuch V beschreibt in §139 Absatz 2, ein Hilfsmittel sei dann ins Hilfsmittelverzeichnis aufzunehmen,
„wenn der Hersteller die Funktionstauglichkeit und Sicherheit, die Erfüllung der Qualitätsanforderungen nach Absatz 2 und, soweit erforderlich, den medizinischen Nutzen nachgewiesen hat und es mit den für eine ordnungsgemäße und sichere Handhabung erforderlichen Informationen in deutscher Sprache versehen ist.“
Auf dieser Basis hat der GKV-Spitzenverband konkrete, Produktgruppen-übergreifende Anforderungen in seiner Verfahrensordnung zur Erstellung und Fortschreibung des Hilfsmittelverzeichnisses definiert:
6.1 Anforderung: Funktionstauglichkeit und Sicherheit
Bei zugelassenen Medizinprodukten gelten die Funktionstauglichkeit und die Sicherheit bereits mit der CE-Kennzeichnung als nachgewiesen, auch wenn zusätzliche Prüfungen in Verdachtsfällen möglich sind.
Die Definition von Medizinprodukten für das Hilfsmittelverzeichnis (HMV) bezieht sich auf das Medizinproduktegesetz (MPG) und nicht die Medical Device Regulation (MDR). Der Grund dafür liegt darin, dass die MDR auch Produkte einschließt, die dem Begriff der Arzneimittel zugeordnet werden könnten, was für Hilfsmittel einen zu weit gefassten Rahmen darstellt. Allerdings gelten für den Nachweis der Funktionstauglichkeit und Sicherheit bei Aufnahmeanträgen die Regelungen der MDR, einschließlich der dort beschriebenen Konformitätsbewertungsverfahren.
Für Hilfsmittel, die keine Medizinprodukte darstellen, erstellt der GKV-Spitzenverband konkrete Anforderungen an die Funktionstauglichkeit und Sicherheit in den einzelnen Produktuntergruppen des HMV. Diese finden sich in den Dokumenten zu den Fortschreibungen der Produktgruppen.
Ein Hilfsmittel muss also nicht zwingend auch ein zertifiziertes MDR-Medizinprodukt sein.
6.2 Anforderung: Besondere Qualitätsanforderungen
Die besonderen Qualitätsanforderungen werden Indikations- oder einsatzbezogen festgelegt. Der GKV legt entsprechende Qualitätsanforderungen und das Nähere zum Nachweis der Erfüllung dieser Anforderungen in den Produktuntergruppen des HMV (ggf. mit der Beschreibung von Prüfverfahren) und den dazu gehörenden Antragsformularen fest. Die genauen Anforderungen finden sich ebenfalls zu jeder Produkt(unter-)gruppe auf der Webseite des GKV.
6.3 Anforderung: Medizinischer Nutzen
Der Nachweis des medizinischen Nutzens eines Hilfsmittels ist gemäß § 139 SGB V erforderlich, wenn das Hilfsmittel nicht nur dem Behinderungsausgleich dient, sondern auch im Rahmen einer ärztlich verantworteten Krankenbehandlung eingesetzt werden soll.
Falls das Hilfsmittel untrennbar mit einer neuen, in der ambulanten Versorgung noch nicht anerkannten Behandlungsmethode verbunden ist, darf es erst ins HMV aufgenommen werden, wenn der Gemeinsame Bundesausschuss (G-BA) die Methode positiv bewertet hat.
Gelangt der GKV-Spitzenverband zu der Einschätzung, dass das Hilfsmittel als untrennbarer Bestandteil einer neuen Methode anzusehen ist, teilt er das dem Hersteller mit und veranlasst gleichzeitig eine Auskunft durch den G-BA.
Wenn das Hilfsmittel bewährte Methoden verwendet, müssen wissenschaftliche Studien seinen medizinischen Nutzen bestätigen – gemäß des “aktuellen, allgemein anerkannten Stands der medizinischen Erkenntnisse”. Eine einzelne Studie reicht nicht. Die Methode muss sich etabliert haben.
6.4 Anforderung: Gebrauchsanweisung
HMV-Produkte müssen Informationen auf Deutsch enthalten, die eine sichere und korrekte Nutzung gewährleisten. Diese Angaben – je nach Produktgruppe im HMV spezifiziert – umfassen Anwendungshinweise, Indikationen, Risiken, Kontraindikationen sowie Anweisungen zu Betrieb, Reinigung, Desinfektion, Montage und Material, sofern der Hersteller diese Anforderungen nachgewiesen hat.
7. Antrag zur Aufnahme ins Hilfsmittelverzeichnis
Schauen wir uns jetzt, wie viel Zeit der Prozess zur Aufnahme in das Hilfsmittelverzeichnis in Anspruch nimmt – und wie die einzelnen Schritte konkret aussehen:
- Antragstellung: Der Antrag auf Aufnahme muss vom Hersteller oder einem beauftragten Dritten gestellt werden. Dieser Antrag kann online über die Plattform des GKV-Spitzenverbands eingereicht werden. Davor sollte selbstverständlich sichergestellt sein, dass die nötigen Anforderungen (siehe obige Kapitel) erfüllt sind.
- Prüfung: Der GKV-Spitzenverband erteilt dem Antragsteller innerhalb von zehn Werktagen nach Antragseingang eine Eingangsbestätigung und stellt innerhalb von zehn Wochen fest, ob der Antrag formal vollständig ist. Dabei handelt es sich allein um die formale Feststellung der Vollständigkeit, nicht um die inhaltliche Vollständigkeit des Antrags. Werden unvollständige Unterlagen eingereicht, gibt es eine Frist von bis zu sechs Monaten, um diese nachzureichen.
- Entscheidung: Wird alles fristgerecht eingereicht, entscheidet der GKV innerhalb von drei Monaten über den Antrag und informiert den Antragsteller über den Ausgang des Verfahrens.
- Erfolg der Aufnahme: Wenn das Produkt aufgenommen wird, erhält es eine individuelle Nummer und wird im Hilfsmittelverzeichnis sowie im Bundesanzeiger veröffentlicht.
Der gesamte Prozess dauert aufgrund der komplexen Bewertung und eventuell notwendigen Rückfragen durchschnittlich zwischen sechs und zwölf Monaten.
8. Vergütung und Preisfindung mit den Krankenkassen
Die Aufnahme eines Produkts ins Hilfsmittelverzeichnis (HMV) ist ein wichtiger Schritt, garantiert jedoch noch keine automatische Erstattung. Bevor Versicherte das Produkt über die Krankenkassen beziehen können, muss die Vergütungshöhe geklärt werden. Hersteller haben verschiedene Optionen, um dies zu erreichen.
8.1 Beitritt zu existierenden Verträgen
Für viele Produktgruppen existieren bereits Rahmenverträge zwischen Krankenkassen oder deren Verbänden und den Hilfsmittel-Herstellern. Als Hersteller eines Hilfsmittels haben Sie die Möglichkeit, diesen Verträgen beizutreten, wenn ein entsprechender Vertrag für Ihre Produktgruppe verfügbar ist.
Einerseits ermöglicht diese Option Ihnen einen schnellen Marktzugang, da keine neuen Verhandlungen mit einzelnen Krankenkassen erforderlich sind. Die Rahmenverträge regeln die Details der Versorgung, einschließlich der Vergütung. Auf der anderen Seite könnten die festgelegten Vergütungssätze in den Verträgen für Ihr Produkt ggf. unpassend bzw. zu niedrig sein.
Eine Anlaufstelle hierfür ist der Verband der Ersatzkassen e. V. (vdek). Hersteller können den bereits verhandelten Verträgen des vdek beitreten. Mehr Informationen bezüglich Verträgen des vdek finden Sie hier.
8.2 Einzelvereinbarungen zur Vergütung
Wenn keine passenden Verträge existieren oder Sie spezielle Konditionen benötigen, können individuelle Einzelvereinbarungen mit den Krankenkassen getroffen werden. Diese Option bietet mehr Flexibilität, erfordert jedoch einen erheblichen Zeitaufwand. Jede Krankenkasse muss einzeln kontaktiert und überzeugt werden.
Einzelvereinbarungen sind vor allem dann sinnvoll, wenn Ihr Produkt einzigartig ist und nicht in eine bestehende Produktgruppe mit Rahmenverträgen passt. Gemäß § 127 Abs. 3 SGB V sind Krankenkassen verpflichtet, individuelle Vereinbarungen zu treffen, wenn keine passenden Verträge bestehen und eine Versorgung der Versicherten anders nicht möglich ist.
8.3 Festbeträge für Produktgruppen
Für viele Hilfsmittel legt der GKV-Spitzenverband Festbeträge fest, welche den maximalen Erstattungsbetrag definieren. Trotz dieser Festbeträge müssen Hersteller Verträge mit den Krankenkassen oder deren Verbänden abschließen bzw. bestehenden Verträgen beitreten, um Ihre Produkte an Versicherte abgeben zu können. Die Festbeträge dienen dabei als Höchstgrenze für die Vergütung. Es ist allerdings auch möglich, niedrigere Vergütungen zu vereinbaren, um bspw. im Preiswettbewerb einen Vorteil zu erlangen.
Festbeträge setzen neben der Obergrenze für die Leistungen der gesetzlichen Krankenversicherung gleichermaßen den maximalen Anspruch der Versicherten auf Versorgung. Möchten Versicherte eine Versorgung in Anspruch nehmen, die über den Festbetrag hinausgeht, müssen sie die Differenz aus eigener Tasche zahlen. Die definierten Festbeträge finden sich auf der Webseite des GKV.
9. Fortschreibung der Produktgruppen
Alle 5 Jahre wird die grundsätzliche Fortschreibung der Produktgruppe (und das Anforderungsprofil) überprüft.
Das bedeutet für die Hersteller: Der GKV-Spitzenverband kann zum Zweck der Fortschreibung von Ihnen die „zur Prüfung der Anforderungen des HMV erforderlichen Unterlagen“ erneut einfordern.
Der GKV-Spitzenverband teilt Ihnen hierzu in einem Schreiben die Nachweisunterlagen mit, die benötigt werden, und bis zu welcher Frist diese einzureichen sind.
Sollte diese Prüfung nicht erfolgreich für Sie verlaufen, wird Ihr Produkt gemeinsam mit der Erstattungsfähigkeit aus dem Hilfsmittelverzeichnis entfernt.
10. Beispiele: Gelistete Software-Produkten im Hilfsmittelverzeichnis
Es ist gar nicht so leicht, Standalone Software-Hilfsmittel – also Software, die ohne die Kopplung an ein spezifisches, physisches Gerät auskommt, im Hilfsmittelverzeichnis zu finden.
In der Produktgruppe 16 „Kommunikationshilfen“ wird man jedoch fündig. Speziell die Untergruppe 5 „Behinderungsgerechte Software für Kommunikationssysteme“ ist hier interessant. Hier finden sich derzeit (Stand: 2. Dezember 2024) acht Softwareprodukte:
- OnScreenKeys
- Meine-eigene-Stimme, Version 2+
- OnScreenCommunicator
- WorldWide
- MULTiTEXT ohne Sprachausgabe
- MULTiTEXT mit Sprachausgabe
- MULTiTEXT mit Bildschirmtastatur ohne Sprachausgabe
- MULTiTEXT mit Bildschirmtastatur mit Sprachausgabe
- VISIOcoach
Auch wenn diese Liste nicht vollständig ist: So viel mehr Standalone-Software-Produkte finden sich leider aktuell nicht im Hilfsmittelverzeichnis. Hierbei ist zu bedenken, dass das Hilfsmittelverzeichnis schon deutlich länger als z. B. das DiGA-Verfahren beim BfArM existiert. Dennoch gibt es kaum Standalone-Software oder mobile Apps im Verzeichnis.
Was sich allerdings sehr häufig finden lässt, sind Hardware-Software-Kombinationen. Produkt-Kombinationen, wo z. B. eine mobile App die Steuerung eines elektronischen Geräts übernimmt, gibt es in großen Zahlen im Hilfsmittel-Verzeichnis.
Woran liegt es aber, dass es so wenig Standalone-Software gibt?
11. Warum gibt es kaum Standalone-Software im Hilfsmittelverzeichnis? (Hürden)
Woran liegt es nun also, dass es bisher verhältnismäßig wenig Standalone-Softwareprodukte in das Hilfsmittelverzeichnis geschafft haben?
Grund 1: Fehlende passende Produktgruppen
Oftmals sind in einer Produktuntergruppe definitorisch komplette Systeme – beispielsweise ein iPad inklusive Kommunikationssoftware oder ein Notrufsystem mit Tracker am Handgelenk – gefordert. Das lässt wenig Spielraum, eine reine Softwarelösung in einer Produktgruppe unterzubringen.
Der Standalone-Software-Hersteller müsste also eine neue Produktuntergruppe beantragen. Bis zur erfolgreichen Erstellung dieser Produktuntergruppe können viele Jahre vergehen, was diesen Prozess beispielsweise für Start-ups unpraktikabel macht.
Grund 2: Festgelegte preisliche Höchstbeträge
Die definierten Festbeträge, die ursprünglich für physische Produkte oder traditionelle medizinische Hilfsmittel festgelegt wurden, spiegeln oft nicht die Besonderheiten und den Wert heutiger Softwareprodukte wider. Etwa die hohen Entwicklungskosten, der kontinuierliche Wartungsaufwand oder die laufenden Updates, die bei Software oder mobilen Apps essenziell sind, werden hier nicht abgedeckt und berücksichtigt. Aus diesem Grund kann es für Hersteller von Softwareprodukten unattraktiv oder gar wirtschaftlich unrentabel sein, ihre Produkte in solchen Kategorien listen zu lassen. Die Festbeträge setzen eine Obergrenze für die Erstattung, die möglicherweise deutlich unter den tatsächlichen Kosten oder dem Marktwert der Software liegt.
Grund 3: Aufwändiger Prozess für Einzelvereinbarungen zur Höhe der Erstattungsbeträge
Ohne Beitritt zu einem existierenden Vertrag muss der Hersteller erhebliche zusätzliche Ressourcen in Verhandlungen von Einzelvereinbarungen investieren, was einerseits die finanziellen Kosten erhöht und andererseits den Marktzugang verzögern kann.
In der Praxis zeigt sich außerdem: Die Chancen, eine Einzelvereinbarung mit einer Krankenkasse zu treffen, sofern es geeignete bestehende Verträge für die Produktgruppe gibt, sind äußerst gering. Zudem ist der Ressourcenaufwand für die Verhandlung einer Einzelvereinbarung nicht zu unterschätzen.
Vielen Dank an dieser Stelle an Christoph Müller, Vorstandsvorsitzender des Bundesfachverbands Elektronische Hilfsmittel e.V. (BEH), für die zahlreichen Informationen und das Mitwirken an diesem Fachbeitrag.
12. Weitere Wege in die Kostenerstattung durch Krankenkassen
Neben dem Hilfsmittelverzeichnis gibt es außerdem einige weitere Wege, die Software-Herstellern die Kostenerstattung im deutschen Gesundheitssystem ermöglichen. Dazu gehören unter anderem:
- Überblick über alle Wege in die Kostenerstattung: Zu unserem Whitepaper
- Kostenerstattung für Software im Krankenhaus: Zu unserem Whitepaper
- Digitale Gesundheitsanswendungen – DiGA-Verfahren: Zu unserem Leitfaden
- Selektivverträge mit Krankenkassen: Zu unserem Leitfaden
- Zentrale Prüfstelle Prävention (ZPP): Zu unserem Leitfaden
- Digitale Pflegeanwendungen – DiPA-Verfahren: Zu unserem Leitfaden
- NUB als Kostenerstattung für Software-Produkte im Krankenhaus: Zu unserem Leitfaden
- Finanzierung über den Innovationsfonds: Zu unserem Leitfaden
- Öffentliche Förderungen für Digital Health-Produkte: Zu unserem Leitfaden
13. Fazit: Lohnt sich das Hilfsmittelverzeichnis für Software-Hersteller?
Zusammenfassend lässt sich sagen: Es lohnt sich, als Hersteller einer medizinischen Software einen Blick auf das GKV-Hilfsmittelverzeichnis zu werfen.
Wichtig ist hierbei, dass Ihr Produkt zum Charakter eines Hilfsmittels und gleichzeitig in eine existierende Produktgruppe passt. Außerdem sollten Sie vorab prüfen, ob ggf. Festbeträge für Ihr Produkt gelten und ob diese für Ihr Unternehmen ausreichend sind.
Unter den richtigen Umständen eröffnet sich hier ein geeigneter Erstattungsweg in die Regelversorgung. Dieser Weg ist für geeignete Produkte sogar deutlich schneller als die Aufnahme in das mittlerweile stark regulierte DiGA-Verzeichnis.
DiGA-Hersteller könnten jedoch auf Herausforderungen stoßen, wenn sie eine Listung im HMV anstreben. Der Fokus von DiGA liegt in der Regel auf therapie- und behandlungsorientierten Zielen, während das Hilfsmittelverzeichnis primär auf unterstützende Anwendungen ausgerichtet ist, die Behinderungen oder Einschränkungen ausgleichen. Dieser unterschiedliche Schwerpunkt macht eine Aufnahme ins HMV für DiGA oft wenig naheliegend.
Produkte, bei denen eine Software die Steuerung eines elektronischen, physischen Hilfsmittels übernimmt, sind wiederum gut geeignet, um als Teil des Hilfsmittel-Verzeichnisses erstattet zu werden.
Wenn Sie eine medizinische Software planen und einen Entwicklungspartner suchen, kontaktieren Sie uns gern. Als spezialisierter Dienstleister liegt unser Fokus auf der regulatorisch-konformen Entwicklung von medizinischen Apps und Gesundheitssoftware. Wir helfen Ihnen, ein Produkt zu planen und umzusetzen, das am Ende erstattungsfähig und im Gesundheitsmarkt erfolgreich ist.
The post Erstattung von Software im GKV-Hilfsmittelverzeichnis (HMV) appeared first on QuickBird Medical.
]]>The post DiGA in Belgien – Zulassung digitaler Gesundheitsanwendungen appeared first on QuickBird Medical.
]]>In Belgien wurde neben Deutschland und Frankreich ein spezieller Zulassungsprozess für digitale Gesundheitsanwendungen etabliert. Auch in Österreich gibt es seit 2020 stärker werdende Bestrebungen, DiGA in die reguläre Gesundheitsversorgung zu integrieren. Mit Blick auf die beiden Vorreiterländer wurde das große Potenzial für eine patientenzentrierte medizinische Versorgung festgestellt und daher ein spezieller Zulassungspfad geschaffen: die mHealth-Pyramide. Mit dieser soll der belgischen Bevölkerung der Zugang zu qualitätsgeprüften und wirksamen digitalen Gesundheitsanwendungen gegeben werden.
Wir werfen in diesem Artikel einen detaillierten Blick auf die Schritte des Zulassungsprozesses und Sie erfahren, welche Anforderungen es zu beachten gilt, um erstattungsfähige digitale Gesundheitsanwendungen erfolgreich in Belgien einzuführen.
Darüber hinaus nimmt dieser Artikel auch die Unterschiede und Ähnlichkeiten zwischen dem deutschen DiGA-Fast-Track und der belgischen mHealth-Pyramide unter die Lupe: Durch den Vergleich beider Ansätze erhalten Sie einen umfassenden Überblick über die verschiedenen Wege, die die beiden Länder eingeschlagen haben, um digitale Gesundheitsanwendungen zu fördern.
Ein besonderer Fokus wird dabei auf der Möglichkeit zur vorläufigen Zulassung mit Analogie zum deutschen DiGA-Fast-Track liegen.
Inhaltsverzeichnis
- 1. Zulassung innerhalb der mHealth-Pyramide
- 2. Der belgische Zulassungsprozess für Stufe 3 der mHealth-Pyramide
- 2.1 Kriterien für eine Zulassung auf Stufe 3
- 2.2 Antragsverfahren beim NIHDI/ INAMI
- 2.3 Ablauf der Bewertung zur vorläufigen Zulassung
- 2.4 Ablauf der Bewertung zur dauerhaften Zulassung
- 2.5 Schlüsselaspekte für nicht-belgische Antragsteller
- 2.6 Wer darf einen Antrag stellen?
- 2.7 Welche digitalen Gesundheitsanwendungen werden bisher vom NIHDI erstattet?
- 3. Übersicht: mHealth Stufe 3 versus DiGA-Fast-Track
- 4. DiGA-Modelle in anderen Ländern
- 5. Fazit
1. Zulassung innerhalb der mHealth-Pyramide
Zuerst werfen wir einen Blick auf die mHealth-Pyramide, um eine grobe Übersicht über das belgische Konzept hinsichtlich Struktur, Verantwortlichkeiten und Anforderungen zu erhalten.
Sie bildet seit 2021 den Zulassungsprozess für vielfältige digitale Anwendungen ab, von der Videosprechstunde bis zu digitalen Gesundheitsanwendungen. Der offizielle Titel dieser digitalen Lösungen ist „Applications de santé mobiles“ oder „Applications mobiles médicales“. Dabei handelt es sich nicht ausschließlich um Apps für die Nutzung auf Smartphones. Auch reine Desktop-Anwendungen für medizinisches Fachpersonal führt mHealthBELGIUM in der Liste der Softwarelösungen auf.
1.1 Aktueller Stand des belgischen Zulassungsprozesses
Die mHealth-Pyramide und der damit verbundene rechtliche Rahmen für die Finanzierung von medizinischen Apps wurde 2016 entwickelt. Beides wurde im Januar 2021 abgeschlossen und liegt als königlicher Erlass vor. Da es sich noch um ein neues Verfahren im belgischen Gesundheitssystem handelt, sind in Zukunft noch Anpassungen erwarten.
Eine Liste der digitalen Gesundheitsanwendungen findet sich auf der Website von mHealthBELGIUM. Das Verzeichnis wird allerdings nur in unregelmäßigen Abständen aktualisiert.
1.2 Aufbau der mHealth-Pyramide
Die mHealth-Pyramide in Belgien ist in drei Stufen mit zunehmend strengeren Anforderungen aufgebaut und erfordert die Interaktion mit unterschiedlichen öffentlichen Stellen. Dieses Stufensystem wurde für ein breites Spektrum digitaler Gesundheitslösungen konzipiert.
Die untenstehende Abbildung der Pyramide soll eine erste Orientierung für die darauffolgende Erläuterung geben. Auf der linken Seite sind in groben Zügen die Anforderungen ausgewiesen, die auf der jeweiligen Stufe erfüllt werden müssen. Auf der rechten Seite sind die involvierten Institutionen aufgeführt.
Von besonderem Interesse sind im weiteren Verlauf des Artikels die blau hervorgehobenen Stufen „3-“ und „3+“: Sofern eine digitale Gesundheitsanwendung die Anforderungen dieser Stufen erfüllt, wird sie von den belgischen Krankenkassen erstattet.
 Zulassung im Rahmen der mHealth-Pyramide in Belgien
Zulassung im Rahmen der mHealth-Pyramide in Belgien
1.3 Das Stufensystem der mHealth-Pyramide
Eine digitale Anwendung muss, sobald sie ein CE-Kennzeichen als Medizinprodukt trägt, verpflichtend auf einer der Stufen der mHealth-Pyramide zugelassen werden, um in Belgien auf den Markt gebracht zu werden. Die grundlegende klinische Wirksamkeit und Sicherheit sind also bereits an diesem Punkt gesichert.
Eigenschaften und Zweck Ihres Produkts sowie Ihr Zielvorhaben mit der Zulassung beeinflusst die Stufe, die Sie wählen:
- Für digitale Gesundheitsanwendungen, die lediglich Gesundheitsdaten sammeln und an medizinisches Fachpersonal übertragen, reicht dabei eine Zulassung auf Stufe 1.
- Sobald die Anwendung eine Funktion zur aktiven Überwachung von Gesundheitszuständen oder Diagnosefunktionen aufweist, muss die Anwendung die Kriterien der Stufe 2 erfüllen.
- Eine Zulassung auf Stufe 3 ist dann stets freiwillig und wird vom Antragsteller nur angestrebt, wenn die Gesundheitsanwendung von den belgischen Krankenkassen erstattet werden soll.
Da die Stufen aufeinander aufbauen, werden sie, falls erforderlich, nacheinander durchlaufen: Die Nachweise aus einer Stufe werden für die Zulassung auf der nächsten Stufe benötigt.
Mehr Informationen zu den einzelnen Stufen finden Sie in der folgenden Tabelle:
| Zulassungsbedingung | zuständige Stelle |
Vergütungsmöglichkeit
|
|
| Stufe 1 |
|
FAHMP (Bundesagentur für Arzneimittel und Gesundheitsprodukte) |
individuelle Vereinbarungen mit Kliniken oder Krankenkassen |
| Stufe 2 |
|
FAHMP (Bundesagentur für Arzneimittel und Gesundheitsprodukte) Koordination über die sogenannte eHealth-Plattform an der verschiedene Institutionen mitwirken |
individuelle Vereinbarungen mit Kliniken oder Krankenkassen |
| Stufe 3 |
|
NIHDI (Nationales Institut für Kranken- und Invalidenversicherung) |
Vergütung durch die Krankenkassen (vorläufig oder dauerhaft)
|
2. Der belgische Zulassungsprozess für Stufe 3 der mHealth-Pyramide
Im Folgenden konzentrieren wir uns auf die Anforderungen für die Zulassung einer digitalen Gesundheitsanwendung auf Stufe 3. Sowohl die Analogien als auch die Unterschiede zum deutschen DiGA-Fast-Track-Verfahren werden hier deutlich. Untenstehend finden Sie außerdem einen tabellarischen Vergleich zwischen dem deutschen und belgischen Prozess für die Zulassung digitaler Gesundheitsanwendungen.
2.1 Kriterien für eine Zulassung auf Stufe 3
Zuständig für den Antragsprozess ist das NIHDI. Es heißt mit vollem Namen Nationales Institut für Kranken- und Invalidenversicherung und hat eine Vielzahl an Aufgaben im belgischen Gesundheitssystem. Es firmiert auch unter der französischen Abkürzung INAMI.
Entscheidend für die Finanzierung digitaler Gesundheitsanwendungen durch die belgischen Krankenkassen sind folgende Punkte:
- Erfüllung der Kriterien aus Stufe 1 und 2
- Integration in ein Versorgungskonzept: Schlüsselelement, um den gesundheitsökonomischen Mehrwert nachzuweisen
- Klinische Wirksamkeit
- Gesundheitsökonomischer Mehrwert: Die App führt beispielsweise zu Verbesserungen in der Patientenversorgung, Kosteneinsparungen oder anderen Vorteilen.
Für eine vorläufige Zulassung auf Stufe „3-„ müssen die Nachweise aus den Punkten 2. bis 4. noch nicht erbracht sein. Es müssen jedoch Nachweiskonzepte mit den Antragsunterlagen eingereicht werden. Dies kann zum Beispiel ein Studiendesign sein.
2.2 Antragsverfahren beim NIHDI/ INAMI
Antragsteller entscheiden sich zuerst, ob sie eine vorläufige oder direkt eine dauerhafte Erstattung durch das NIHDI beantragen möchten und stellen das Dossier mit den notwendigen Unterlagen dementsprechend zusammen.
Ähnlich wie im deutschen BfArM kommt zur Bewertung des Antrags ein multidisziplinärer Ausschuss zusammen. Derzeit umfasst das Gremium 13 Personen aus verschiedenen Interessensgruppen wie Krankenkassen, Patientenvertretern oder Mitgliedern des NIHDI.
2.3 Ablauf der Bewertung zur vorläufigen Zulassung
Der Ausschuss bewertet den eingegangenen Antrag auf Vollständigkeit und berät in üblicherweise drei Sitzungen über die Möglichkeit der vorläufigen Erstattung.
Möglichkeiten zur Ablehnung bestehen an allen Stellen des Verfahrens:
- wenn die Antragsunterlagen nicht vollständig sind.
- wenn die vorläufige Erstattung abgelehnt wird.
- wenn die eingereichten Nachweise für eine dauerhafte Erstattung als ungenügend angesehen werden.
Hersteller haben während der Entscheidungsfindung des Gremiums die Möglichkeit, das Antragsdossier persönlich zu erläutern und auf Nachfragen zu reagieren.

Belgien – Der Weg von der vorläufigen zur dauerhaften Erstattung durch das NIHDI
Die Dauer der vorläufigen Zulassung beträgt üblicherweise drei Jahre. In dieser Zeit wird die Gesundheitsanwendung bereits von den Krankenkassen erstattet. Der Preis wird vom Antragsteller mit dem NIHDI verhandelt.
Zum Vergleich: In Deutschland beträgt die standardmäßige vorläufige Erstattungsdauer nur 1 Jahr. Außerdem wird der Preis während der vorläufigen Zulassung in Deutschland vom Hersteller festgelegt. Erst wenn die DiGA dauerhaft erstattet werden soll erfolgt eine Preisverhandlung zwischen Hersteller und GKV-Spitzenverband.
Während der Phase der vorläufigen Erstattung muss der Antragsteller nach 18 Monaten an das NIHDI über den Fortschritt des Umsetzungsplans zur endgültigen Zulassung berichten. Die vollständigen Unterlagen für den Entscheid über die dauerhafte Zulassung und Erstattung müssen spätestens 6 Monate vor dem Ende des Erprobungszeitraumes vorliegen.
2.4 Ablauf der Bewertung zur dauerhaften Zulassung
Entscheiden sich Antragsteller direkt die dauerhafte Zulassung zu beantragen, so werden von Anfang an alle Unterlagen von einer Arbeitsgruppe des NIHDI auf Vollständigkeit geprüft und dem Antragsteller am Ende ein Bescheid erteilt.
Dies bedeutet, dass sämtliche Nachweise über
- die klinische Wirksamkeit,
- den positiven sozioökonomischen Effekt,
- und die erfolgreiche Einbindung in einen Versorgungsprozess
schon vor Antragstellung vollständig vorliegen müssen.
In Belgien ist zum Veröffentlichungszeitpunkt dieses Blog-Artikels noch kein direkter Antrag auf dauerhafte Zulassung gestellt worden. Praxisbeispiele dazu fehlen also bisher.
2.5 Schlüsselaspekte für nicht-belgische Antragsteller
Für eine erfolgreiche Zulassung einer digitalen Gesundheitsanwendung beim NIHDI ist ein fundiertes Verständnis des belgischen Gesundheitssystems von Vorteil, da einige Besonderheiten zu beachten sind. Im Vergleich mit dem deutschen DiGA-Fast-Track Verfahren fallen diese Unterschiede auf, die sich explizit auf die Einbettung der Argumentation in den Kontext des belgischen Gesundheitssystems beziehen:
- Integration in einen Versorgungspfad
Im Gegensatz zu deutschen DiGA können digitale Gesundheitsanwendungen immer nur als integriert in einen innovativen Versorgungspfad zugelassen und auch vergütet werden. Dieses Kriterium hat keine exakte Entsprechung im deutschen DiGA-Fast-Track.
Diese Integration gelingt nicht ohne Partnerschaften im belgischen Gesundheitssystem. Hersteller können mit Gesundheitseinrichtungen, wie Krankenhäusern und Arztpraxen, zusammenarbeiten, um die Anwendung in bestehende Versorgungsprozesse zu integrieren. Pilotstudien sind eine Möglichkeit, die erfolgreiche Integration nachzuweisen. Über User-Feedback oder Erfahrungsberichte von medizinischem Fachpersonal können die Vorteile der Behandlung dokumentiert werden. Gegebenenfalls können auch Aspekte einer klinischen Studie die gelungene Integration in die belgische Versorgungslandschaft nachweisen.
Für die Erfüllung dieses zusätzlichen Kriteriums sind gute Kontakte zu Akteuren im belgischen Gesundheitssystem wichtig.
- Klinische Wirksamkeit
Für den Zulassungsantrag in Belgien kann der Nachweis der klinischen Wirksamkeit durch eine Studie aus dem Ausland, wie beispielsweise aus Deutschland für eine DiGA, verwendet werden. Das Produkt hat bereits durch die CE-Kennzeichnung als Medizinprodukt Nachweise für Sicherheit und Wirksamkeit erbracht. Es muss jedoch unter anderem plausibel begründet werden, warum dieser Nachweis auch für das belgische Gesundheitssystem gültig ist. Eine Bezugnahme auf die belgische Population und der Nachweis der Übertragbarkeit der Studienergebnisse sind unerlässlich.
- Gesundheitsökonomischer Effekt:
Auch beim Nachweis des gesundheitsökonomischen Effekts gelten ähnliche Bedingungen. Während in Deutschland der Fokus bei DiGA stark auf der Verbesserung der Patientenversorgung liegt, betont Belgien zusätzlich die Bedeutung von Prozessverbesserungen im Gesundheitswesen. Ein Beispiel dafür könnte die Verringerung postoperativer Komplikationen oder eine effizientere Patientenbetreuung sein.
Argumente aus bestehenden Unterlagen einer DiGA-Zulassung in Deutschland sind nur hilfreich, wenn sie nachweisen können das das vorgestellte Versorgungskonzept sowohl innovativ als auch den bestehenden belgischen Konzepten überlegen ist.
2.6 Wer darf einen Antrag stellen?
Seit Oktober 2023 haben nicht nur die Hersteller, sondern auch verschiedene andere Akteure in Belgien die Möglichkeit, in Zusammenarbeit mit den Herstellern Anträge für die Zulassung digitaler Gesundheitsanwendungen zu stellen. Dazu zählen Vertriebsunternehmen, Krankenhäuser, wissenschaftliche Verbände oder Berufsverbände.
Beispielsweise können Kliniken, die innovative Versorgungskonzepte einführen wollen und dabei auf digitale Gesundheitsanwendungen als Bausteine setzen, ebenfalls Anträge beim NIHDI stellen.
Beim NIHDI stehen die jeweiligen Antragsformulare zur Verfügung.
Beispiele aus der Praxis fehlen hierfür bisher. Die erweiterte Möglichkeit zur Antragstellung wird nach einem Jahr vom NIHDI bewertet. Es ist daher in Zukunft mit Anpassungen zu rechnen.
2.7 Welche digitalen Gesundheitsanwendungen werden bisher vom NIHDI erstattet?
Zum Veröffentlichungszeitpunkt dieses Blog-Artikels werden lediglich die drei Anwendungen des Herstellers moveUP erstattet.
Eine davon ist die Smartphone-App moveUP, die zur Rehabilitation nach Knie- oder Hüftoperationen verwendet wird. Sie schlägt aufgrund der Informationen personalisierte Übungen vor und überträgt die von den Patienten eingegebenen Informationen an die behandelnden Ärzte.
Am Beispiel von moveUP wird deutlich, wie bedeutend die Integration in einen Versorgungspfad und der daraus resultierende sozioökonomische Nutzen sind.
moveUP wurde von Anfang an nicht isoliert entwickelt, sondern als integraler Bestandteil eines Versorgungspfades, der sowohl für die Klinik als auch für die Patienten Vorteile bietet.
- Smartphone App für Patienten: In der Smartphone-App moveUP erhalten Patienten auf sie abgestimmte Übungen für die Rehabilitation nach einer Knie- oder Hüft-OP. Außerdem beantworten sie Fragen zu ihrem Gesundheitszustand. Die Daten werden an die behandelnden Ärzte übermittelt.
- Desktopanwendung für Ärzte: Auf ärztlicher Seite steht eine Desktopanwendung zur Übertragung und Auswertung der Daten zur Verfügung, die die Patienten in der Smartphone-App move Up erfassen.
- Coaching- System: Es gibt in der mobilen App eine Anbindung an ein Coaching-System, in welchem den Patienten Fragen direkt durch medizinisches Personal beantwortet werden. Dadurch muss die Arztpraxis nicht für kleinere Anliegen aufgesucht werden.
Durch die Bereitstellung dieser drei Anbindung entsteht der neue Versorgungspfad.
3. Übersicht: mHealth Stufe 3 versus DiGA-Fast-Track
Für einen besseren Überblick werden hier noch einmal die wesentlichen Elemente der Zulassungswege in Belgien und Deutschland miteinander verglichen.
Ähnlichkeiten der Stufe 3 der mHealth Pyramide mit dem deutschen DiGA-Fast-Track Verfahren
| Stufe 3 der mHealth-Pyramide |
Deutscher DiGA-Fast-Track
|
|
| zuständige Behörde |
NIHDI |
BfArM |
| Zulassungssystem |
Stufe 3 mit Ähnlichkeiten zum deutschen DiGA-Fast-Track |
DiGA Fast-Track-Prozess |
| Zielgruppe |
Apps mit therapeutischer und/oder diagnostischer Funktion |
Apps mit therapeutischer und/oder diagnostischer Funktion |
| Möglichkeit zur vorläufigen Zulassung |
ja üblicherweise für 3 Jahre |
ja für 1 Jahr, Verlängerung um 1 Jahr möglich in Einzelfällen |
| Nachweis positiver Versorgungseffekt |
verpflichtend für dauerhafte Zulassung |
verpflichtend für dauerhafte Zulassung |
| Erstattungsmechanismus |
durch die Krankenkassen |
durch die Krankenkassen |
Unterschiede der Stufe 3 der mHealth Pyramide mit dem deutschen DiGA-Fast-Track Verfahren
| Stufe 3 der mHealth-Pyramide |
Deutscher DiGA-Fast-Track
|
|
| mögliche Antragsteller |
|
Hersteller |
| zugelassene Risikoklassen nach MDR |
alle |
I-IIa (zukünftig auch IIb) |
| Preisverhandlungen |
|
|
| Nachweis sozioökonomischer Effekt |
vorgeschrieben |
nein |
| Integration in bestehende Versorgungspfade |
vorgeschrieben |
nein Eine DiGA kann alleinstehend verordnet werden. |
| Rezept notwendig für die Nutzung der App? |
teilweise |
ja |
| Verzeichnis aller zugelassenen Apps |
inaktuelles Verzeichnis von mHealthBELGIUM |
DiGA-Verzeichnis des BfArM |
4. DiGA-Modelle in anderen Ländern
Immer mehr Länder planen einen standardisierten Prozess, um digitale Gesundheitsanwendungen in die Regelversorgung zu bringen. Neben Belgien sollten Sie außerdem einen Blick in die folgenden Länder werfen:
- DiGA in Deutschland
- DiGA in Frankreich
- DiGA in der Schweiz
- DiGA in Österreich
- DiGA in Italien
- Überblick über Zulassung von DiGA in allen Ländern der EU
Außerdem gibt es eine breite Palette von öffentlichen Förderungen (siehe unser Praxisleitfaden zu Digital Health-Förderungen) und anderen Erstattungswegen (siehe unser Whitepaper zu Selektivverträgen) für Health-Software.
5. Fazit
Belgien zeigt sich in vielen Bereichen des Gesundheitssystems sehr aufgeschlossen gegenüber der Digitalisierung, was sich unter anderem in der Existenz der elektronischen Patientenakte und der Gewöhnung der Bürger an digitale Versorgungsprozesse widerspiegelt.
Dies schafft für Hersteller digitaler Gesundheitsanwendungen einen attraktiven Markt in einem digitalisierungsfreundlichen Umfeld.
Die pragmatische Herangehensweise der belgischen Behörden bei der Zulassung von Gesundheitsapps kann sowohl vorteilhaft als auch herausfordernd sein. Einerseits ist somit in kurzer Zeit ein neuer Zulassungsprozess geschaffen worden. Andererseits sind einige Anforderungen zum aktuellen Zeitpunkt noch nicht klar definiert und es ist mit weiteren Anpassungen zu rechnen. Diese dynamische Situation sorgt für Unsicherheit bei Herstellern.
Wenn Sie die Zulassung einer digitalen Gesundheitsanwendung in Belgien planen, kontaktieren Sie uns gerne. Wir helfen Ihnen die Anwendung zu entwickeln und den optimalen Zulassungspfad zu finden.
The post DiGA in Belgien – Zulassung digitaler Gesundheitsanwendungen appeared first on QuickBird Medical.
]]>The post Formulierung der Zweckbestimmung für (Software-)Medizinprodukte appeared first on QuickBird Medical.
]]>In diesem Artikel erhalten Sie eine praktische Anleitung zur Erstellung Ihrer Zweckbestimmung nach MDR. Dabei fokussieren wir uns auf Softwareprodukte, wie mobile Apps und Webanwendungen. Die Konzepte und Definitionen sind aber auf jedes andere Medizinprodukt übertragbar.
Inhalt
- 1. Mit 3 Fragen zur Zweckbestimmung
- 2. Zweckbestimmung – das Fundament Ihres Medizinprodukts
- 3. Fazit
Bevor Sie sich Hals über Kopf in Ihr neues Projekt stürzen, ein Entwicklerteam zusammenstellen und schon über die Vermarktung nachdenken, lassen Sie uns einen Schritt zurückgehen und von vorne beginnen: bei der Zweckbestimmung. Denn erst, wenn diese steht, wissen Sie, welche regulatorischen Anforderungen auf Sie zukommen und wie Sie Ihr Produkt am Ende bewerben dürfen.
Die Zweckbestimmung legt das Fundament für (fast) alles. Ganz zu Beginn eines Software-Projekts lässt sich damit nämlich die wohl wichtigste Frage überhaupt beantworten: ist Ihre Software ein Medizinprodukt und müssen Sie sich daher an die Vorgaben der Medical Device Regulation (MDR) halten?
Beispiel: Angenommen, Sie wollen eine App entwickeln, die die körperliche Beweglichkeit von Menschen steigert, indem Sie basierend auf Nutzereingaben ein individuelles Trainingsprogramm erstellt. Die Trainingsinhalte bestehen aus Informationen über den Bewegungsapparat und diversen Dehnübungen. Eine mögliche (sehr vereinfachte) Zweckbestimmung könnte lauten:
Die App ist dazu bestimmt, die Beweglichkeit von gesunden Erwachsenen zu steigern.
In diesem Fall würde sich die Software nicht als Medizinprodukt qualifizieren, da sie nicht der Definition der MDR entspricht. Grob zusammengefasst besagt diese nämlich, dass nur Apps, die einen Zweck in Zusammenhang mit einer Krankheit oder Behinderung verfolgen, als Medizinprodukt gelten. Dazu zählen unter anderem Diagnose, Therapie und Kompensierung (die genaue Definition finden Sie in unserem Artikel zum Thema: Leitfaden: Ist Ihre App ein Medizinprodukt?). Demnach ändert sich die Situation für Ihre Software, wenn Sie die Zweckbestimmung leicht abändern:
Die App ist dazu bestimmt, die Beweglichkeit von Menschen nach einer Beinamputation zu steigern und Strategien zur Kompensation der Behinderung aufzubauen.
So schnell kann es gehen! Durch die Veränderung eines einzelnen Satzes haben Sie Ihre App zu einem Medizinprodukt gemacht. Dazu waren nicht einmal Änderungen an der Software notwendig.
1. Mit 3 Fragen zur Zweckbestimmung
Um eine Zweckbestimmung zu schreiben, benötigen Sie keine jahrelange Erfahrung im regulatorischen Bereich. Mit genügend Expertise in Bezug auf Ihr Produkt und Ihre Zielgruppe können Sie diese auch ohne große Vorkenntnisse allein verfassen. Lassen Sie ihre Zweckbestimmung aber am besten am Ende noch einmal von einem Regulatorik-Experten überprüfen.
Die MDR definiert die Zweckbestimmung folgendermaßen:
„Zweckbestimmung“ bezeichnet die Verwendung, für die ein Produkt entsprechend den Angaben des Herstellers auf der Kennzeichnung, in der Gebrauchsanweisung oder dem Werbe- oder Verkaufsmaterial bzw. den Werbe- oder Verkaufsangaben und seinen Angaben bei der klinischen Bewertung bestimmt ist;
Diese Definition lässt drei Fragen ableiten, welche für die Inhalte der Zweckbestimmung zentral sind:
- “Was soll auf meiner Produkt-Webseite stehen?”: Versetzen Sie sich in die Lage Ihres Endkunden und überlegen Sie sich, welche Informationen er benötigt, um eindeutig zu wissen, dass Ihr Produkt für ihn geeignet ist und welchen Mehrwert es ihm liefern kann.
- “Welche Versprechen kann das Produkt auch halten?”: Leere Versprechungen sind tabu. Daher dürfen Sie auch nur jene Punkte nennen, die Sie innerhalb der klinischen Bewertung auch nachweisen können.
- „Was kann das Produkt nicht?“ (falls notwendig): Manchmal ist es auch notwendig, die Grenzen des Produkts zu thematisieren. Darunter zählt zum Beispiel der Hinweis, dass ein Produkt nur einmal zu verwenden ist.
1.1 Was die Zweckbestimmung nicht enthält
Die MDR versteht die Zweckbestimmung als Teil der Produktbeschreibung und Spezifikation (MDR Artikel 1, Anhang II). Zusätzlich zur Zweckbestimmung enthält diese noch weitere Punkte, die eng miteinander verwandt sind:
- die vorgesehene Patientengruppe (z.B. ICD-Codes, demografische Daten, Kontraindikationen)
- die Grundsätze betreffend den Betrieb des Produkts und seine Wirkungsweise (Beschreibung der Funktionalitäten und wie der Zweck erreicht werden soll)

Was die Zweckbestimmung enthält – und was sie nicht enthält
Viele Hersteller vermischen diese Punkte miteinander. Das passiert auch zwangsläufig, da eine strikte Trennung dieser Themen nicht möglich ist. Dennoch sollte die Zweckbestimmung nicht allzu detailliert auf die Patientengruppe und die Funktionsweise des Produkts eingehen.
Bei der Unterscheidung können folgende Fragen helfen:
- Zweckbestimmung: WAS soll das Produkt bezwecken?
- Patientengruppe: WER soll das Produkt verwenden?
- Grundsätze betreffend des Betriebs des Produkts und seiner Wirkungsweise: WIE funktioniert das Produkt und wie soll es seinen Zweck erfüllen?
1.2 Wie lang muss eine Zweckbestimmung sein?
Ist es falsch, die Patientengruppe und die Funktionalitäten der Software bereits in der Zweckbestimmung detailliert zu erklären? Nein, aber unter Umständen unklug. Denn eine Zweckbestimmung kann im Nachhinein nicht mehr ohne weiteres geändert werden. Grundsätzlich gilt: Die Zweckbestimmung sollte so präzise wie möglich geschrieben sein. Die Schwierigkeit besteht darin, relevante Informationen von irrelevanten zu trennen.
Denn bestimmt möchten Sie Ihr Produkt nach Markteintritt noch weiterentwickeln und um Features ergänzen. Vielleicht sind gewisse Anpassungen sogar notwendig, um die Produktsicherheit zu erhöhen. In diesen Fällen kann es problematisch sein, wenn Ihre Zweckbestimmung die Funktionsweise der App schon im Detail erklärt. Das Gleiche gilt, wenn Sie Ihre Patientenpopulation im Nachgang erweitern wollen oder gezwungen sind, diese einzugrenzen.
Es ist also eine Kunst, alle relevanten Aspekte in der Zweckbestimmung zu erwähnen und dabei nicht zu weit auszuholen.
Hinweis: Bei der Formulierung der Zweckbestimmung gibt es nicht einen einzigen richtigen Weg. Die Zweckbestimmung Ihres Produkts kann viele Formen haben, welche potenziell zulässig sind.
Die Länge der Zweckbestimmung ist sehr produktabhängig. Sie ist so lang wie nötig, um Fehlinterpretationen auszuschließen, sollte das Produkt aber auch nicht zu detailliert beschreiben. Manche Zweckbestimmungen beinhalten nur zwei Sätze, andere sind weitaus detaillierter. An dieser Stelle helfen bestimmt ein paar Beispiele von existierenden Medizinprodukten, um Ihnen eine Orientierung zu geben.
Beispiele
Zweckbestimmung für Skalpellklingen:
- „Sterile Skalpellklingen sind dazu vorgesehen, in aufbereitete Skalpellgriffe aufgenommen zu werden. Sterile Skalpelle sind für Hautoberflächenschnitte (Hautinzision) am menschlichen Körper bestimmt.“
Zweckbestimmung für einen Ballonkatheter:
- „Ballonkatheter sind zur routinemäßigen oder postoperativen Drainage der Blase vorgesehen. Der Einsatz von Ballonkathetern ist nur für urologische Zwecke geeignet. Die Produkte sind nur für den Einmalgebrauch zugelassen und die empfohlene maximale Verweildauer für transurethrale Silikonkatheter beträgt 30 Tage. Die Anwendung darf nur durch qualifiziertes Fachpersonal erfolgen.“
Zweckbestimmungen für DiGA:
- Die Zweckbestimmungen aller aktuell gelisteten DiGA, welche ebenfalls Medizinprodukte darstellen, finden Sie im Verzeichnis des BfArM. Wenn Sie sich unsicher sind, ob es sich bei Ihrer App um eine DiGA handelt, finden Sie hier eine Antwort: Ist Ihre App eine DiGA? Definition & Kriterien digitaler Gesundheitsanwendungen
2. Zweckbestimmung – das Fundament Ihres Medizinprodukts
In der MDR gibt es keine konkrete Liste mit Inhalten, welche in der Zweckbestimmung angeführt werden sollen. Jedoch wird die Zweckbestimmung in vielen Kapiteln erwähnt und als Grundlage für zahlreiche Aktivitäten innerhalb des Softwarelebenszyklus aufgeführt.

Einflussbereiche der Zweckbestimmung nach MDR (kein Anspruch auf Vollständigkeit)
Im Folgenden sind zentrale Punkte aufgeführt, welche von der Zweckbestimmung beeinflusst werden und somit bei ihrer Formulierung berücksichtigt werden müssen.
2.1 Qualifizierung als Medizinprodukt
Die Zweckbestimmung muss klar erkennen lassen, dass das Produkt einen medizinischen Zweck hat. Gemäß der Definition der MDR zählen dazu mitunter die Diagnose, Überwachung und Vorhersage von Krankheiten, sowie die Kompensation von Behinderungen. Die genaue Definition finden Sie in diesem Artikel: Leitfaden: Ist Ihre App ein Medizinprodukt?
2.2 Risikoklassifizierung des Medizinprodukts
Nachdem Sie das Produkt als Medizinprodukt qualifiziert haben, folgt die Evaluierung der Risikoklasse. Konkrete Informationen zur richtigen Risikoklasse finden Sie in diesem Artikel.
Um die Risikoklasse möglichst eindeutig bestimmen zu können, muss die Zweckbestimmung entsprechend präzise formuliert sein. Auch Ausschlüsse (Zwecke, für die das Produkt nicht vorgesehen ist), können, wenn notwendig, angegeben werden. Dies ist der Fall, wenn eine andere Interpretation der Zweckbestimmung zu einer höheren Risikoklasse führen könnte.
Beispiel für einen Ausschluss: Das Produkt liefert keinerlei Informationen an den behandelnden Arzt und stellt kein diagnostisches Instrument dar.
Daher kann es auch sein, dass Sie auf bestimmte Funktionalitäten der Software eingehen müssen, wenn diese die Risikoklasse des Produkts beeinflussen. Sollten Fehlinterpretationen möglich sein, welche die Risikoklasse verändern, sind Diskussionen mit den benannten Stellen oder Aufsichtsbehörden vorprogrammiert.
Hinweis: Wenn Sie eine Digitale Gesundheitsanwendung (DiGA) entwickeln möchten, darf Ihr Produkt nur in Klasse I, IIa oder IIb fallen. Achten Sie also darauf, dass das Produkt gemäß seiner Zweckbestimmung nicht in Klasse III fallen kann. Weitere Informationen zu DiGA finden zum Beispiel hier: Leitfaden zur DiGAV: Digitale-Gesundheitsanwendungen-Verordnung
2.3 Risikoanalyse
Im Zuge des Risikomanagements müssen Sie eine Risikoanalyse für Ihr Produkt durchführen. Auch dafür liefert die Zweckbestimmung maßgeblichen Input. Zentral ist die Frage, wie kritisch die Aufgaben sind, die Ihr Produkt wahrnimmt und wie sensibel das Anwendungsgebiet ist. Wenn Ihre App beispielsweise zur Früherkennung von Herzinfarkten bestimmt ist, können bei Fehlfunktionen oder Falschanwendungen gravierende Schäden entstehen. Während von einer App zur Steigerung der Konzentrationsfähigkeit bei ADHS-PatientInnen weniger große Schäden zu erwarten sind.
Die möglichen Schäden sollten sich aus der Zweckbestimmung ableiten lassen, um in der Risikoanalyse evaluiert werden zu können.
Software Safety Class
Wenn Sie ein Software-Produkt planen, werden Sie sich zwangsläufig auch mit der IEC 62304 auseinandersetzen müssen. Mit dieser Norm lässt sich Ihre Anwendung einer Software Safety Class zuordnen. Die grundlegende Frage dabei lautet:
„Wie groß ist der Schaden, der eintreten kann, wenn Ihre Software eine Fehlfunktion aufweist oder sogar komplett versagt?“
Die Software Safety Class beschreibt unter anderem, wie granular Ihre Software während des Entwicklungsprozesses getestet werden muss und kann demnach zusätzlichen Aufwand bedeuten. Da die Software Safety Class oft aus der Risikoanalyse abgeleitet wird, hängt auch diese Einstufung mit Ihrer Zweckbestimmung zusammen.
2.4 Klinische Bewertung und Post-Market-Surveillance
Nur, wenn ein Produkt auch eine klare Zweckbestimmung hat, kann im Zuge einer klinischen Bewertung gezeigt werden, dass sie diesen Zweck auch erfüllt. Die Zweckbestimmung legt also den Grundstein für die Sicherheits- und Leistungsanforderungen, welche im Zuge der klinischen Bewertung berücksichtigt werden. In manchen Fällen wollen Hersteller den Aufwand einer klinischen Prüfung umgehen und berufen sich daher auf „gleichartige Produkte“. Ein zentraler Punkt für die Gleichartigkeit zweier Produkte ist eine identische Zweckbestimmung (MDCG 2020-5). Hier erfahren Sie mehr über die klinische Bewertung und die Kriterien für die Gleichartigkeit von Produkten: MDR-Guide: Klinische Bewertung von Software-Medizinprodukten
Für mehr Informationen zur Validierung im Allgemeinen sollten Sie unseren Artikel zu diesem Thema lesen: Validierung von Medizinprodukt-Software nach MDR
Wenn eine klinische Prüfung geplant ist, ist es auch von Ihrem Budget abhängig, welche Effekte nachgewiesen werden sollen. In der Zweckbestimmung dürfen nur Leistungsmerkmale angegeben werden, für die es auch einen Nachweis gibt.
Auch in der Post-Market-Surveillance Phase werden Daten über das Produkt gesammelt. Welche Daten dabei zu berücksichtigen sind, wird von der Zweckbestimmung abgeleitet (z.B. Lebensqualität, Dauer einer Reha, etc.).
3. Fazit
Die Zweckbestimmung ist das zentrale Element bei der Entwicklung einer Software als Medizinprodukt. Sie legt unter anderem den Grundstein für folgende Punkte:
- Qualifikation als Medizinprodukt
- Risikoklasse & Software Safety Class
- Risikokanalyse
- Klinische Bewertung & Post-Market-Surveillance Daten
Die Zweckbestimmung für Ihre Software sollte möglichst zu Beginn des Entwicklungsprozesses verfasst werden. Dabei sollte darauf geachtet werden, alle notwendigen Informationen zu inkludieren, aber nicht zu tief ins Detail zu gehen.
Falls Sie eine Medical Software oder DiGA planen und Unterstützung bei der Entwicklung benötigen, setzen Sie sich gerne mit uns in Kontakt und wir besprechen das Ganze zusammen unverbindlich. Wir entwickeln Medical Software für Unternehmen und Forschungseinrichtungen auf Auftragsbasis.
Weitere Fragen? Rufen Sie uns gerne an (+49 (0) 89 54998380) oder schreiben uns eine Email ([email protected]).
The post Formulierung der Zweckbestimmung für (Software-)Medizinprodukte appeared first on QuickBird Medical.
]]>The post DiGAV und DVG: wichtige Links appeared first on QuickBird Medical.
]]>DiGAV: Gesetz bzw. Verordnung online und als Download (PDF, HTML, EPUB, XML)
- DiGAV: Verordnung online einsehbar als HTML
- DiGAV: Inhaltsverzeichnis der Verordnung online einsehbar als HTML
- DiGAV als PDF (Quelle: Gesetze im Internet) – Download
- DiGAV als EPUB (Quelle: Gesetze im Internet) – Download
- DiGAV als XML (Quelle: Gesetze im Internet) – Download
Hinweis: Genaugenommen spricht man bei der DiGAV nicht von einem Gesetz, sondern von einer Verordnung.
DVG: Gesetz online und als Download (PDF)
DVPMG: Gesetz online und als Download (PDF)
DigiG: Gesetz online und als Download (PDF)
Weitere nützliche DiGA-Links
- DiGA-Leitfaden des BfArM
- Unser kompakter Leitfaden zur DiGAV
- Offizielle Informationen des BfArM zum Thema DiGA, DVG und DiGAV
- Definition und Kriterien einer DiGA: Ist Ihre App eine DiGA?
- DiGA Verzeichnis für Hersteller
- Inoffizielles DiGA Verzeichnis vom Spitzenverband Digitale Gesundheitsversorgung
- Das Digital-Gesetz (DigiG): Leitfaden für DiGA-Hersteller
Weitere Fragen?
Als Dienstleister für die Entwicklung von DiGA und Medical Apps sind wir in der Umsetzung der aktuellen Gesetzes-Bestimmungen für DiGA sehr erfahren. Falls Sie weitere Fragen haben, melden Sie sich gerne:
Telefon: +49 (0) 89 54998380
Email: [email protected]
The post DiGAV und DVG: wichtige Links appeared first on QuickBird Medical.
]]>The post Künstliche Intelligenz (KI) in Medizinprodukten – MDR Leitfaden (2026) appeared first on QuickBird Medical.
]]>Viele Hersteller sind sich unsicher, inwiefern KI überhaupt in Medizinprodukten eingesetzt werden darf. Aus diesem Grund wollen wir uns in diesem Artikel mit dem Einsatz künstlicher Intelligenz, insbesondere Machine Learning (ML), in regulierten Medizinprodukten befassen.
Wir fokussieren uns in diesem Leitfaden auf die Implikationen der Medical Device Regulation (MDR) für KI-basierte Medizinprodukte.
Dabei wollen wir unter anderem die folgenden Fragen klären:
- Darf ein Medizinprodukt überhaupt KI enthalten? Erhält man hierfür überhaupt eine Zulassung?
- Darf ein KI-Medizinprodukt während der Nutzung im Live-Betrieb kontinuierlich weiterlernen?
- Welche speziellen Gesetze und ISO/IEC Standards sind einzuhalten?
- Was möchte die benannte Stelle in Bezug auf KI-Medizinprodukte sehen?
- Was müssen Sie während der Entwicklung von Künstlicher Intelligenz als Medizinprodukt beachten?
- Können Sie LLMs wie ChatGPT in Ihrem Medizinprodukt nach MDR nutzen?
Zuallererst stellt sich dabei die Frage:
Was macht KI überhaupt so besonders im regulatorischen Sinn?
Wieso sollte man hierfür überhaupt eine Extra-Behandlung im Sinne der Gesetzgebung erwarten?
Inhalte dieses Artikels
- 1. Regulatorische Besonderheiten von künstlicher Intelligenz (KI)
- 2. Zulassung nach MDR: Medizinprodukte mit künstlicher Intelligenz
- 2.1 Anforderungen an die Entwicklung von KI-Medizinprodukten
- 2.2 Was erwartet die benannte Stelle in Bezug auf KI-basierte Medizinprodukte?
- 2.3 Relevante Normen und Leitfäden für künstliche Intelligenz in Medizinprodukten
- 2.4 Welche Machine Learning-Modelle sind als Medizinprodukt zertifizierbar?
- 2.5 Können Sie LLMs wie ChatGPT in Ihrem Medizinprodukt nach MDR nutzen?
- 2.6 Kosten und Aufwand für die Zulassung
- 3. Weitere regulatorische Anforderungen für KI
- 4. Fazit
1. Regulatorische Besonderheiten von künstlicher Intelligenz (KI)
Um die regulatorischen Besonderheiten von KI zu verstehen, sehen wir uns folgendes Beispiel an:
Wir haben eine Diabetes-App, welche auf Basis der Patienten-Daten eine Empfehlung abgibt, wann und wie viel Insulin sich der Diabetes-Patient injizieren sollte. Hierfür werden Daten z. B. über den Blutzucker-Spiegel, das Essens-Tagebuch und sportliche Aktivitäten berücksichtigt.

Beispiel einer KI für Diabetes-Patienten zur Empfehlung der Insulin-Dosierung
Macht es nun einen Unterschied, ob eine KI diese Empfehlung berechnet oder z.B. ein regelbasierter Entscheidungsbaum?
Die Antwort ist leider „Jein“. In der Theorie ist die gesetzliche Grundlage der Medical Device Regulation (MDR) identisch bei klassischer Software und KI-basierter Software.
In der Praxis erwarten Sie bei KI-basierter Software regulatorische Herausforderungen, die durch die technische Natur von KI entstehen. Die wichtigsten Besonderheiten von KI, die im regulatorischen Sinne eine große Rolle spielen, sind die Folgenden:
1.1 Interpretierbarkeit
Eine KI ist meist eine “Blackbox” für den Menschen. Selbst für den Entwickler der KI kann es schwer nachvollziehbar sein, wie genau die künstliche Intelligenz aus den Eingabedaten bestimmte Ausgaben errechnet hat.
 Entscheidungen künstlicher Intelligenz sind für den Menschen oft nicht nachvollziehbar
Entscheidungen künstlicher Intelligenz sind für den Menschen oft nicht nachvollziehbar
Es ist in unserem Diabetes-KI-Szenario z. B. erstmal nicht klar, wie genau die Diabetes-KI die konkrete Insulin-Menge und den optimalen Injektionszeitpunkt berechnet. Somit ist es für den KI-Entwickler auch schwer zu sagen, ob das System wirklich in allen Situationen richtig funktioniert. Das birgt natürlich hohe Risiken für den Diabetes-Patienten, der bei einer Unterdosis oder Überdosis Insulin körperlichen Schaden nehmen und sogar sterben könnte.
Ein einfacher Entscheidungsbaum oder z. B. lineare Regressionsmodelle sind für den Menschen transparent nachvollziehbar. Dementsprechend ist es mit dem nötigen medizinischen Fachwissen deutlich einfacher zu validieren, ob das System richtig funktioniert.
Man könnte sagen, dass der Entwickler von herkömmlicher Software bereits vor der Entwicklung weiß, auf welcher Basis Entscheidungen getroffen werden. Die KI hingegen erarbeitet dieses Entscheidungsmodell erst auf Basis der Daten, mit denen sie gefüttert wird und ist nicht zwangsläufig dazu in der Lage, dem Entwickler, Arzt oder Patienten dieses Modell im Nachhinein zu erklären.
Regulatorische Herausforderung: Bei einer Medizinprodukt-Software nach MDR müssen Sie validieren, dass Ihr System funktioniert und keine inakzeptablen Restrisiken existieren. Das ist regulatorisch gesehen gar nicht so einfach, ohne tiefes Verständnis des Software-Systems. Zur Zulassung Ihres KI-Systems sollten Sie daher Erkenntnisse und Methoden aus den Gebieten der Transparenz, Erklärbarkeit und Interpretierbarkeit von KI-Systemen nutzen.
1.2 Kontinuierliches Lernen
Mit KI und speziell Machine Learning entsteht eine Neuheit, die auch regulatorische Herausforderungen mit sich bringt: Systeme, die selbstständig und kontinuierlich weiter lernen. Manche KI-Systeme werden auch während der Nutzung des Produkts fortlaufend trainiert und ändern somit die eigene Verhaltensweise im Live-Betrieb.
Das fortlaufende Lernen der KI passiert jedoch außerhalb des direkten Einflussbereichs des Entwicklers. Dementsprechend ist es umso schwerer vorherzusehen, ob die KI in Zukunft weiterhin richtig funktioniert.

Beispiel: Microsoft hat im Jahre 2016 einen Twitter-Bot veröffentlicht, der automatisiert Tweets generieren konnte. Dieser hat aktiv auf Basis neuer Daten weitergelernt. Durch die absichtliche Manipulation der Community hat der Twitter-Bot aber leider irgendwann viele rassistische Aussagen gemacht. Ein gutes Beispiel dafür, dass selbstlernende Systeme schwerer kontrollierbar sind.
Zum Vergleich, es gibt natürlich auch die Möglichkeit, KI-Systeme nur initial während der Entwicklung mit Daten zu trainieren und nach Integration in das Produkt „einzufrieren“ (auch „statische“ KI genannt). Wie wir weiter unten im Artikel erläutern, sollte dies die präferierte Variante für die meisten Software-Medizinprodukte im Sinne der regulatorischen Zulassung sein.
Regulatorische Herausforderung: Selbst-lernende KI-Systeme ändern noch nach der initialen regulatorischen Zulassung das eigene Verhalten. Diese Verhaltens-Änderung basiert meist auf nicht-validierten Daten, die von Nutzern bereitgestellt werden. Das bringt regulatorische Herausforderungen für die Validierung und das Change-Management (bzw. Änderungsmanagement) im Rahmen des MDR-Medizinprodukts mit sich.
1.3 Abhängigkeit von Daten
Daten sind für ein KI-Medizinprodukt eine zentrale Ressource. Während herkömmliche Software meist auf Basis vordefinierter Regeln funktioniert, leitet ein Machine Learning Algorithmus diese Regeln von den Daten ab, mit denen er trainiert wird.
Wenn Sie ein neuartiges KI-Modell entwickeln wollen, benötigen Sie also zuallererst Daten. Wenn Sie keinen Zugriff auf verlässliche medizinische Daten haben, müssen Sie diese ggf. innerhalb einer klinischen Studie mühsam selbst generieren.
Für vortrainierte Modelle (wie z. B. GPT-4 von ChatGPT) benötigen Sie zwar keine Trainingsdaten. Diese sind aktuell aber leider aufgrund von sog. „Halluzinationen“ selten verlässlich genug für einen medizinischen Anwendungsfall.
Regulatorische Herausforderung: Zur Zulassung eines verlässlichen KI-Modells benötigen Sie Zugriff auf valide, medizinische Daten. Diese Daten zu erhalten, kann kosten- und zeitintensiv sein.
Im nächsten Kapitel gehen wir darauf ein, wie Sie diese Besonderheiten von Artificial Intelligence (AI) bei der Zulassung Ihres Medizinprodukts nach MDR berücksichtigen sollten.
Anmerkung: Der Einfachheit halber werden in diesem Artikel KI allgemein behandeln. Insgesamt schließt dies aber verschiedene Unterkategorien, wie Generative AI, Large Language Models (LLM) und Machine Learning (ML) mit ein. Diese Begriffe werden an einigen Stellen auch beispielhaft verwendet.
2. Zulassung nach MDR: Medizinprodukte mit künstlicher Intelligenz
Es gibt aktuell (noch) keine gesonderten Anforderungen an Software-Medizinprodukte mit KI-Komponenten (Stand: Januar 2025). In der MDR suchen Sie vergebens nach Begriffen wie KI, AI oder Machine Learning.
Regulatorisch gesehen, sind die wichtigsten Normen für Sie bei Software-basierten Medizinprodukten also weiterhin u.a.:
- ISO 13485: Qualitätsmanagementsystem (zum ISO 13485-Guide)
- IEC 62304: Software-Lebenszyklus-Prozesse (zum IEC 62304-Guide)
- IEC 82304: Anforderungen an Gesundheitssoftware
- IEC 62366-1: Gebrauchstauglichkeit
- ISO 14971: Risikomanagement
Mehr Informationen zur Entwicklung von Software-Medizinprodukten (mit oder ohne künstliche Intelligenz) finden Sie hier.
Aktuell gibt es noch keine etablierte (geschweige denn harmonisierte) Norm speziell für die Entwicklung von KI-basierten Medizinprodukten. Künstliche Intelligenz wird im Sinne der MDR und der oben genannten Normen derzeit genauso geregelt wie klassische Software-Produkte.
Ganz so einfach ist es jedoch nicht:
Sie halten sich an dieselben Normen und Gesetze wie bei klassischer Medizinprodukt-Software. Sie müssen dadurch aber natürlich auch wie bei jedem anderen Medizinprodukt u. a. die folgenden Anforderungen abdecken:
- Sie müssen die Sicherheit des Medizinprodukts im Griff haben: Wie hoch ist das Risiko, dass jemand durch Nutzung Ihres Produkts zu Schaden kommt? Wie können Sie dieses Risiko minimieren? Ist das Restrisiko akzeptabel? Etc.
- Sie müssen die Leistungsfähigkeit Ihres Medizinprodukts sicherstellen und nachweisen können: Kann Ihr Produkt die bezweckte Leistung auch wirklich liefern? Können mit Ihrer Software z.B. wirklich Krankheiten diagnostiziert werden oder basiert Ihre Aussage nur auf Bauchgefühl? Mit welcher Genauigkeit und Fehlerrate kann eine Krankheit diagnostiziert werden? Etc.
Bei künstlicher Intelligenz sind die oben genannten Fragen zur Sicherheit und Leistungsfähigkeit meist deutlich schwerer zu beantworten als bei Software mit transparenten, regelbasierten Algorithmen. Das liegt vorwiegend an den im vorangegangenen Kapitel genannten Besonderheiten von künstlicher Intelligenz. KI ist initial erstmal eine „Blackbox“, und die Sicherheit und Leistungsfähigkeit dieser sind scheinbar schwer bewertbar.
Woher wissen Sie, dass Ihre künstliche Intelligenz in jedem Anwendungskontext richtig funktioniert?

Beispiel: Sie entwickeln eine App, welche Hautkrebs mit Hilfe der Kamera Ihres Smartphones diagnostizieren soll. Angenommen, die Trainingsdaten für Ihre KI sind aus Deutschland und beinhalten somit ggf. mehr Bilder von Personen mit helleren Hauttönen als von Personen mit dunkleren Hauttönen. Können Sie sicherstellen, dass die Vorhersage Ihrer KI für Personen mit dunkleren Hauttönen einwandfrei funktioniert? Außerdem: Wie geht Ihr Algorithmus mit einem Bild um, bei welchen zusätzlich eine Narbe oder ein Feuermal zu sehen ist? Sind Sie sich wirklich sicher, dass solche Fälle ausreichend in den Trainingsdaten der KI repräsentiert sind?
Die Herausforderung liegt hier weniger darin, eine spezielle KI-Norm umzusetzen, sondern im Grunde darin:
„Kennen Sie sich gut genug mit der Entwicklung künstlicher Intelligenz aus, um nachweislich eine sicheres und leistungsfähiges KI-Modell zu entwickeln?“
Und auch wenn Sie die Expertise haben, ist es insbesondere in Deutschland nicht einfach an medizinische Daten für das Training Ihrer KI zu kommen. Die zweite große relevante Frage ist also:
„Haben Sie genug hochqualitative Trainingsdaten für Ihren Anwendungsfall?“
2.1 Anforderungen an die Entwicklung von KI-Medizinprodukten
Wie oben schon beschrieben, gibt es zum aktuellen Zeitpunkt noch keine harmonisierte Norm für MDR-Medizinprodukte, welche konkrete Anforderungen an die Entwicklung von KI-Medizinprodukt definiert. Es gibt jedoch auch jetzt schon Normen und Guidelines, an denen Sie sich orientieren können, um die wichtigen Aspekte abzudecken.
Zur Veranschaulichung der technischen Komplexität haben wir eine beispielhafte Frageliste zusammengestellt. Um sicherzustellen, dass Ihre KI am Ende nicht die falschen Krankheits-Diagnosen stellt oder gefährliche Therapie-Anweisungen gibt, sollten Sie sich z. B. mit folgenden Aspekten befassen:
| Frage | Beispiel für mögliches Problem |
| Woher stammen die Trainingsdaten für Ihre KI? | Ihre Trainingsdaten sind aus einer Quelle, für die Sie die Validität der Daten nicht überprüfen können. Die Daten repräsentieren die reale Welt nicht ausreichend und Ihre KI ist fehlerhaft in der Anwendung. |
| Repräsentieren die Trainingsdaten Ihren Anwendungsfall und die Ziel-Patientengruppe ausreichend? | Ihre Trainingsdaten decken nur einen von vielen Anwendungskontexten ab. Die KI funktioniert nur einem Kontext und schlägt in anderen Situationen fehl. |
| Wie definieren Sie die „Wahrheit“ („Ground Truth“) im Rahmen Ihres Machine Learning-Modells? Woher stammen die Labelling-Daten für Ihr Modell und können Sie der Wahrheit dieser Labels vertrauen? | Die Labels für Ihre Trainingsdaten wurden von einer einzigen Person nach subjektiver Einschätzung vergeben. Andere Personen hätten für die gleichen Daten unterschiedliche Labels vergeben. Die Labels repräsentieren keine fundamentale Wahrheit. |
| Wurde Ihr Modell mit Daten validiert, die nicht zuvor für das Training und Testing verwendet wurden? | Ihr KI wurde mit denselben Daten validiert, die auch im Training und Testing verwendet wurden. Die Validierung ist demnach erfolgreich für die Trainingsdaten, die KI ist aber fehlerhaft für komplett neue Daten im Live-Betrieb des Produkts (auch „Overfitting“ genannt). |
| Haben Sie die Ziel-Patientengruppe und Ziel-Nutzungsumgebung ausreichend charakterisiert und eingegrenzt?
|
Ihre Trainingsdaten decken nur einen von vielen Anwendungsbereiche Ihres KI-Produkts ab. Die KI funktioniert also nur in den Szenarien, für die Sie auch trainiert wurde, aber nicht in anderen Szenarien, die jedoch Teil des echten Anwendungsbereichs sind. |
| Wie reagiert Ihr Modell auf Daten, die es während des Trainings nicht gesehen hat, insbesondere auf seltene oder atypische Fälle? | Ihre KI könnte bei seltenen Krankheitsbildern oder atypischen Patientendaten falsche Diagnosen stellen oder ungeeignete Therapien vorschlagen, da sie für solche Fälle nicht angemessen trainiert wurde. |
| Wie robust ist Ihr Modell gegenüber Manipulationen oder externen Angriffen, insbesondere wenn es um die Sicherheit der Patientendaten geht? | Anfälligkeiten für Cyberangriffe könnten nicht nur die Funktionsfähigkeit Ihrer KI beeinträchtigen, sondern auch sensible Patienteninformationen gefährden. |
| Haben Sie Methoden verwendet, die es ermöglichen, die Entscheidungen Ihrer KI zu erklären und nachzuvollziehen? | Ohne Erklärbarkeit könnte es schwierig sein zu verstehen, wie die KI zu einer bestimmten Diagnose oder Therapieempfehlung gekommen ist. Ihr KI-Modell könnte fehlerhaft sein, ohne dass Sie davon wissen. |
| Enthalten Ihre Daten alle notwendigen Merkmale (Features) und können Sie die Auswahl der observierten Merkmale begründen? | Ihre Trainingsdaten enthalten nicht alle relevanten Merkmale, die für eine präzise Diagnose oder Therapieempfehlung notwendig sind. |
| Etc. | Etc. |
Dies sind nur einige Beispiele zur Veranschaulichung der Komplexität und die Liste könnte ohne Probleme noch um viele Punkte erweitert werden. Eine gute Liste relevanter Fragen finden Sie auch im neuen Fragenkatalog für benannte Stellen zum Thema künstliche Intelligenz in Medizinprodukten. Das führt uns zum nächsten Punkt:
2.2 Was erwartet die benannte Stelle in Bezug auf KI-basierte Medizinprodukte?
Ein Blick in den aktuellen Fragenkatalog für benannte Stellen für KI-basierte Medizinprodukte lohnt sich.
Hier finden Sie die aktuellen Versionen zum Download: Fragebogen der benannten Stellen
(Tipp: Holen Sie sich einen Kaffee und warten einige Minuten, bis der Download startet. Scheinbar sind die Server der IG-NB durchgehend überlastet.)
 Fragebogen der benannten Stellen zu künstlicher Intelligenz
Fragebogen der benannten Stellen zu künstlicher Intelligenz
Sie sollten damit rechnen, im Audit Ihres Medizinprodukts mit derartigen Fragen der benannten Stelle konfrontiert zu werden. Sie benötigen hier jeweils eine schlüssige Antwort, die sich auch in der technischen Dokumentation Ihres Medizinprodukts wiederfindet.
Falls Sie ein KI-Software-Produkt entwickeln, das unter die Risikoklasse I nach MDR fällt, erwartet Sie zumindest unter MDR kein Audit der benannten Stelle (eine benannte Stelle könnte aber für das Konformitätsbewertungsverfahren für den AI-Act für KI-Produkte notwendig sein. Lesen Sie mehr darüber in diesem Artikel.). Sie werden jedoch zu unangekündigten Zeitpunkten von Ihrer Aufsichtsbehörde geprüft. Auch hier lohnt sich ein Blick in den Fragenkatalog.
Benötigen Sie Unterstützung bei der Entwicklung eines medizinischen KI-Systems?
Wir entwickeln medizinische KI-Systeme für Unternehmen im Gesundheitswesen auf Auftragsbasis.
Unsere KI-Services:
– Technischer Review von medizinischen KI-Systemen
– Entwicklung von neuen Machine Learning-Modellen im Gesundheitsbereich
– Individuelle Beratung bezüglich regulatorischer Compliance für Ihr geplantes KI-basiertes Produkt
2.3 Relevante Normen und Leitfäden für künstliche Intelligenz in Medizinprodukten
Wie oben schon erwähnt, erwarten Sie im Rahmen der MDR erstmal keine konkreten neuen gesetzlichen Verpflichtungen für KI-Produkte.
Provokativ gesagt: Wenn Sie diese klassischen Normen für die Entwicklung von medizinischer Software akribisch einhalten, brauchen Sie keine AI-Norm.
Die praktischen Implikationen der MDR für die Umsetzung der KI ergeben sich indirekt u. a. aus:
- … den Kontrollmaßnahmen für Risiken der KI in Bezug auf die menschliche Gesundheit (ISO 14971).
- … der verpflichtenden Verifizierung und Validierung des KI-Systems (ISO 13485, IEC 62304).
In der Praxis ist es aber aus den folgenden Gründen ratsam, sich mit bestimmten KI-spezifischen Normen und Leitlinien zu beschäftigen:
- Hilfestellung für die Entwicklung: Die Entwicklung von verlässlichen KI-Algorithmen ist komplex. Wenn Sie in diesem Bereich nicht absoluter Experte sind, hilft ein Leitfaden, um alle technischen Risiken von KI zu identifizieren und effektiv abzuwenden.
- Interpretationsspielraum der MDR und Medizinprodukt-Normen: Verschiedene Prüfstellen haben andere Interpretationen der MDR-Anforderungen und Normen. Ein Blick in den Fragebogen der benannten Stellen hilft Ihnen dabei, die Erwartungshaltung Ihres zukünftigen Auditors besser zu verstehen.
- Argumentationsbasis im Audit: Ihr Auditor wird wissen wollen, wie Sie sicherstellen, dass Ihre KI verlässlich funktioniert. Wenn Sie darauf antworten „Ich bin KI-Experte und weiß sowas einfach“ ist das für Auditoren eine deutlich weniger zufriedenstellende Antwort als „Wir haben uns bei der Entwicklung und Validierung an die folgenden Normen und Leitlinien gehalten.“. Die öffentliche Akzeptanz einer Norm oder Leitlinie schafft Vertrauen in der Kommunikation mit dem Auditor.
Einige KI-spezifische Leitlinien und Normen, die für Sie in diesem Rahmen relevant sein könnten, sind die z.B. Folgenden:
- Leitfäden der FDA zum Einsatz von Machine Learning in Software as a Medical (SaMD):
- Fragebogen der benannten Stelle
- Leitfaden zur KI bei Medizinprodukten von Christian Johner, Christoph Molnar et al.
- BS/AAMI 34971:2023, Application of ISO 14971 to machine learning in artificial intelligence
- ISO/IEC TR 24028:2020, Edition 1.0 – Information technology – Artificial intelligence – Overview of trustworthiness in artificial intelligence
- ISO/IEC TS 4213:2022, Information technology – Artificial intelligence – Assessment of machine learning classification performance
- ISO/IEC FDIS 23894 Information technology — Artificial intelligence — Guidance on risk management
- ISO/IEC 5338 Information technology — Artificial intelligence — AI system life cycle processes
- ISO/IEC FDIS 5339 Information Technology – Artificial Intelligence – Guidance for AI applications
- ISO/IEC DIS 5259-2 – Artificial intelligence — Data quality for analytics and machine learning (ML) — Part 2: Data quality measures
- ISO/IEC DIS 5259-3 – Artificial intelligence — Data quality for analytics and machine learning (ML) — Part 3: Data quality management requirements and guidelines
- ISO/IEC 5259-4 – Artificial intelligence — Data quality for analytics and machine learning (ML) — Part 4: Data quality process framework
Wir hoffen, dass sich in naher Zukunft eine Norm (oder mehrere) für die Entwicklung von KI-basierten Medizinprodukten etabliert und harmonisiert wird. Ein etablierter Standard hilft sowohl den Firmen bei der Entwicklung als auch den benannten Stellen bei der Auditierung. Wann dies passiert, ist aber bisher nicht absehbar.
2.4 Welche Machine Learning-Modelle sind als Medizinprodukt zertifizierbar?
Bei der Beantwortung dieser Frage scheinen sich selbst die benannten Stellen schwer zu tun. In einer früheren Version des Fragebogens für benannte Stellen (Version 4, vom 09.06.2022) stand noch explizit:
- “Static locked AI” ist zertifizierbar (Produkte mit abgeschlossenem Lernprozess und nachvollziehbaren Entscheidungen)
- “Static black box AI” ist im Einzelfall zertifizierbar (Produkte mit abgeschlossenem Lernprozess und nicht bzw. nur teilweise nachvollziehbaren Entscheidungen)
- “Continuous learning AI” ist nicht zertifizierbar (Produkte, deren Lernprozess im Feld weitergeht)
Im neuen Fragebogen (Version 1, vom 14.11.2024), in dessen Erstellung nun auch das Team NB – The European Association of Medical devices Notified Bodies eingebunden ist, scheint diese klare Vorgabe aber abgeschwächt worden zu sein. Gerade für letztere gibt es in der Theorie nun doch Möglichkeiten einer Zertifizierung, sofern geeignete Maßnahmen vom Hersteller etabliert wurden.
„Die Praxis hat gezeigt, dass es für die Hersteller schwierig ist, die Konformität von KI-Geräten, die die zugrunde liegenden Modelle durch selbstlernende Mechanismen im Feld aktualisieren, ausreichend nachzuweisen. Derzeit betrachten die benannten Stellen Medizinprodukte, die auf solchen Modellen basieren, nicht als „zertifizierbar“, es sei denn, der Hersteller ergreift Maßnahmen zur Gewährleistung des sicheren Betriebs des Produkts im Rahmen der in der technischen Dokumentation beschriebenen Validierung.“
(Übersetzt aus dem Englischen)
Was genau diese “Maßnahmen zur Gewährleistung des sicheren Betriebs des Produkts” in der Praxis aber sind, bleibt offen. Auch wenn die ursprüngliche Aussage, dass eine Zertifizierung von „Continuous learning AI“ unmöglich ist, entfernt wurde, ist immer noch nicht klar definiert, in welchen Fällen eine Ausnahme gemacht werden kann. In der Praxis hängt dies auch stark mit der Risikoklasse des Produkts zusammen. Je nach Ausmaß des möglichen Schadens bei Fehlverhalten der KI, können Restrisiken durch selbst-lernende Systeme akzeptiert werden oder nicht.

Machine Learning-Modell: Statisch vs. Adaptiv (bzw. „selbstlernend“)
Wenn Sie die Entwicklung eines Continuous Learning Systems planen, sollten Sie auf jeden Fall definieren, in welchem Ausmaß der Algorithmus im Feld weiterlernen darf.
Beispiel: Hier muss man dann auch im Rahmen des Risikomanagements analysieren, welche Art von Risikokontrollmaßnahmen man einführen könnte. Bei einer Insulin-Pumpe könnte man z.B. sagen, dass Minimum und Maximum der Insulin-Dosis fest definiert werden und nur die Optimierung dazwischen auf KI basiert.
Daumenregel: Grundsätzlich gilt die Daumenregel, ein einfacheres (interpretierbareres) ML-Modell einer “Black Box” vorzuziehen, und statische KI einem selbstlernenden KI-System vorzuziehen. Sprechen Sie Ihren Plan ggf. mit Ihrer benannten Stelle ab (für Risikoklasse IIa oder höher). Der Interpretationsspielraum der Gesetzgebung ist groß und für unterschiedliche Auditoren müssen Sie daher auch mit einer jeweils unterschiedlichen Erwartungshaltung rechnen.

Bild: Stark vereinfachte Daumenregel: Welche Art von KI sollten Sie wählen?
2.5 Können Sie LLMs wie ChatGPT in Ihrem Medizinprodukt nach MDR nutzen?
Generative AI und speziell Large Language Models (LLMs) haben großes Potenzial in der Medizin. Zu den aktuell bekanntesten LLMs gehören z. B.:
- GPT-5.2 von OpenAI (wird in ChatGPT genutzt)
- Llama von Meta
- Claude von Anthropic
- Speziell im medizinischen Bereich: z. B. Med-PaLM von Google
Der große Vorteil von diesen LLMs:
- Es handelt sich um vortrainierte Modelle. Sie müssen das KI-Modell nicht selbst trainieren und benötigen somit auch keine Trainingsdaten.
- Die Fähigkeiten dieser vortrainierten Modelle sind vielfältig. Durch den Einsatz von LLMs werden viele Innovationen in der Medizin möglich, die bisher nicht vorstellbar waren.
Können Sie Generative AI und speziell LLMs also regulatorisch gesehen in Ihrem Medizinprodukt nutzen?
Regulatorisch gesehen gibt es hier keinerlei explizite Einschränkungen. Sie können potenziell jedes Machine Learning-Modell verwenden, solange Sie sicherstellen, dass das Produkt sicher ist und seinen medizinischen Zweck erfüllt.
Leider sind LLMs aktuell noch nicht verlässlich genug für viele medizinische Anwendungsfälle (Stichwort: „Halluzinationen“). Dadurch ergibt sich bei vielen Einsatzgebieten ein nicht akzeptables Risiko für Ihr Medizinprodukt nach MDR. Für weniger kritische Einsatzgebiete ist es aber nicht explizit ausgeschlossen, dass Sie ein LLM in Ihr Medizinprodukt zu integrieren. Dies muss jeweils im Einzelfall betrachtet werden.
In Zukunft werden LLMs sicherlich kontinuierlich weiter verbessert, was viele neue Einsatzgebiete in der Medizin ermöglichen wird.
2.6 Kosten und Aufwand für die Zulassung
Faktor 1: Risiken Ihres Medizinprodukts
Wie präzise Sie bei der Entwicklung und Validierung Ihres KI-Modells arbeiten müssen, hängt stark von der Höhe der Risiken Ihrer Medizinprodukt-Software ab. Je höher das Risiko Ihres Produkts, desto aufwendiger und kostenintensiver wird die Zulassung.
💡 Beispiel eines hohen Risikos:
Angenommen, Sie entwickeln einen KI-Algorithmus, welcher die optimale Strahlungsdosis für ein Bestrahlungsgerät berechnen soll. Wenn Ihr KI-System einen Fehler macht, kann der Krebs-Patient im schlimmsten Fall durch eine Strahlungs-Überdosis sterben. Dementsprechend viel Aufwand müssen Sie bei der Entwicklung und Validierung Ihrer Medizinprodukt-Software investieren, um die Wahrscheinlichkeit eines Fehlverhaltens nahezu gegen Null laufen lassen.
💡 Beispiel eines niedrigen Risikos:
Angenommen, Sie entwickeln eine digitale Gesundheitsanwendung für die Behandlung von Adipositas bzw. Übergewicht. Ihr KI-System soll nun je nach Tageszeitpunkt und Stimmung des Nutzers, die optimale Fitnessübung vorschlagen. Wenn Ihr Algorithmus ab und zu mal eine nicht ganz optimale Empfehlung abgibt, kommt nicht direkt jemand zu Schaden. Entsprechend haben Sie bei der Entwicklung des KI-Systems die Möglichkeit eine gewissen Fehlerquote als Restrisiko zu akzeptieren. Der Aufwand für die Entwicklung und Validierung des Systems ist überschaubar.
Daumenregel:
Sie benötigen deutlich mehr Daten und müssen mehr Zeit in die Entwicklung Ihres AI-Modells stecken, wenn Ihr Produkt in einer höheren Risikoklasse nach MDR und einer höheren Sicherheitsklasse nach IEC 62304 angesiedelt ist.
Faktor 2: Nutzen Ihres Medizinprodukts
Nach MDR muss ein Medizinprodukt die Risiken immer dem Nutzen gegenüberstellen. Ein neuartiges Medizinprodukt mit hohem Nutzen kann also unter Umständen auch größere Risiken akzeptieren.
Ein KI-Produkt mit seltenen Fehlern, welches durch den Einsatz deutlich mehr Menschenleben rettet als durch Nicht-Anwendung des Medizinprodukts, kann potenziell trotzdem zugelassen werden.
Wenn Ihr Produkt jedoch mit klassischen, regelbasierten Algorithmen genauso zuverlässig funktionieren würde, sollten Sie Abstand von KI nehmen. Sie haben die Pflicht, alle Risiken Ihres Medizinprodukts so weit wie möglich zu reduzieren. Da durch KI viele neue Risiken entstehen, ist ein einfacher, transparenter Algorithmus ist Zweifelsfall vorzuziehen.
3. Weitere regulatorische Anforderungen für KI
Neben der Medical Device Regulation (MDR), betreffen Sie ggf. noch andere gesetzliche Vorgaben:
- Datenschutz-Grundverordnung (DSGVO): Die DSGVO geht zwar nicht speziell auf KI in ihrem Wortlaut ein, hat aber durchaus Implikationen für diese. Wichtigste Implikationen der DSGVO entstehen z. B. durch die Anforderung von „technischen und organisatorischen Maßnahmen“ (TOM) zum Schutz von personenbezogenen Daten. In Bezug auf Cyber Security ergeben sich z. B. spezielle Herausforderungen für KI-Systeme.
- Artificial Intelligence Act (AI Act) der Europäischen Union: Der AI Act reguliert den Einsatz von KI innerhalb der Europäischen Union. Da der EU-AI Act keine Verordnung speziell für Medizinprodukte ist und eine Erklärung dessen den Rahmen des Artikels sprengen würde, gehen wir in diesem Artikel nicht näher auf diese Neuregelung ein. Sie finden hier einen detaillierten Leitfaden zum AI Act speziell für Medizinprodukt-Hersteller.
Wir beschränken uns in diesem Leitfaden auf die Gesetzgebungen, die speziell für KI größere Auswirkungen haben können. Natürlich gibt es eine Vielzahl weiterer Gesetze, die für jedes Software-Produkt gelten.
4. Fazit
Künstliche Intelligenz ist natürlich auch für regulierte Medizinprodukte nach MDR zulassungsfähig. Wie viel Aufwand und Kosten damit einhergehen, hängt vom Risiko und Nutzen Ihrer Medizinprodukt-Software ab.
Grundsätzlich sollten Sie sich überlegen, ob der Einsatz von künstlicher Intelligenz Ihr Produkt in Bezug auf die Zweckbestimmung signifikant besser macht. Wenn Sie mit einem transparenten, regelbasierten Algorithmus ähnliche Ergebnisse erzielen können, sollten Sie ggf. auf KI verzichten. Der Aufwand ist es für Ihr Unternehmen womöglich nicht wert und die benannte Stelle wird die Entscheidung für ein KI-basiertes System hier ggf. auch anzweifeln.
Wenn Ihr KI-Algorithmus hingegen Mehrwerte schafft, die mit klassischen Algorithmen nicht erreichbar sind, dann möchten wir Ihnen an dieser Stelle Mut zusprechen.
In den USA sind Stand Januar 2025 bereits über 1.000 medizinische KI-Systeme zugelassen worden und auch in Europa erhalten regelmäßig neue Firmen die Zulassung für ihr KI-Systems nach MDR. KI in der Radiologie ist zum Beispiel schon längst keine Neuheit mehr und erreicht in diesem Bereich Dinge, die mit regelbasierten Algorithmen schlichtweg nicht möglich wären.
Benötigen Sie Unterstützung bei der Entwicklung eines medizinischen KI-Systems?
Wir entwickeln medizinische KI-Systeme für Unternehmen im Gesundheitswesen auf Auftragsbasis.
Unsere KI-Services:
– Technischer Review von medizinischen KI-Systemen
– Entwicklung von neuen Machine Learning-Modellen im Gesundheitsbereich
– Individuelle Beratung bezüglich regulatorischer Compliance für Ihr geplantes KI-basiertes Produkt
Ob als Funktion innerhalb von digitale Gesundheitsanwendungen (DiGA) oder als Decision Support Systeme für Ärzte, wir entwickeln passende Machine Learning-Modelle für Ihren Anwendungsfall. Falls Sie ein KI-basiertes Medizinprodukt planen, kontaktieren Sie uns gern. Wir beraten Sie individuell zu Ihrem geplanten Produkt und setzen bei Bedarf das gesamte Software-System für Sie um.
The post Künstliche Intelligenz (KI) in Medizinprodukten – MDR Leitfaden (2026) appeared first on QuickBird Medical.
]]>The post Das Digital-Gesetz (DigiG): Leitfaden für DiGA-Hersteller appeared first on QuickBird Medical.
]]>Überblick
- 1. Das Digital-Gesetz: DigiG- Hintergründe und Ziele
- 2. Welche Neuerungen ergeben sich für DiGA Hersteller?
- 2.1 Aufnahme von DiGA der Risikoklasse IIb
- 2.2 Formulierung zum Versorgungsanspruch Schwangerer
- 2.3 Preisgestaltung anhand von Erfolgsmessungen
- 2.4 Keine 14-tägige Rückgabefrist
- 2.5 Zusendung von Freischaltcodes innerhalb von 2 Tagen durch die Krankenkassen
- 2.6 Interoperabilität
- 2.7 Telemedizinisches Monitoring
- 2.8 Innovationsfonds
- 2.9 Strukturierte Behandlungsprogramme bei Diabetes
- 2.10 Schreiben der DiGA in die ePA und Implementierung der GesundheitsID
- 2.11 Einführung und Weiterentwicklung des E-Rezepts
- 3. Der Weg des DigiG durch die Beschlussgremien
- 4. Die Stellungnahmen der Interessengruppen zum Entwurf des DigiG
- 5. Fazit
1. Das Digital-Gesetz: DigiG- Hintergründe und Ziele
Das Digital-Gesetz (DigiG) ist das Gesetz zur Beschleunigung der Digitalisierung im Gesundheitswesen. Es wurde im Februar 2024 im Bundesrat final verabschiedet, um die zahlreichen Potenziale der digitalen Transformation in der Gesundheits- und Pflegebranche zu nutzen. Das DigiG hat das Ziel, das Gesundheitswesen effizienter, patientenzentrierter, sicherer und qualitativ hochwertiger zu gestalten und den Fortschritt in der Versorgung voranzutreiben. Es adressiert die Herausforderungen, die mit der Einführung digitaler Lösungen im Gesundheitswesen einhergehen. Unter anderem zielt das Gesetz darauf ab, die elektronische Patientenakte flächendeckend durch eine Opt-out-Lösung zu integrieren und das E-Rezept verbindlich weiterzuentwickeln, um die Patientensicherheit und die Versorgungsqualität zu verbessern.
2. Welche Neuerungen ergeben sich für DiGA Hersteller?
2.1 Aufnahme von DiGA der Risikoklasse IIb
DiGA der Risikoklasse I und IIa können bereits seit der Verabschiedung des Digitale-Versorgung-Gesetzes (DVG) im Dezember 2019 im Rahmen der Regelversorgung verordnet werden. Neu ist, dass laut Digital-Gesetz auch DiGA der Risikoklasse IIb zugelassen werden können. Hersteller können allerdings keine vorläufige Zulassung beantragen, sondern müssen den positiven Versorgungsnachweis direkt bei Antragstellung auf Aufnahme in das DiGA-Verzeichnis mitreichen. Dies bedeutet, dass eine DiGA nicht schon während der Prüfung zum positiven Versorgungseffekt verordnet werden kann, wie es andernfalls im Fast-Track-Verfahren vorgesehen wäre (vorläufige Listung). Für Hersteller ergeben sich somit längere Zeiträume, die bis zur Umsatzgenerierung überbrückt werden müssen.
2.2 Formulierung zum Versorgungsanspruch Schwangerer
Im DigiG ist die Versorgung von Schwangeren mit DiGA nun ausdrücklich aufgeführt. Notwendig war dies, da eine regelhaft verlaufende Schwangerschaft als ICD-10 Code an sich keine Krankheit darstellt. Trotzdem ist im SGB V die medizinische Versorgung rund um Schwangerschaft, Geburt und Wochenbett geregelt. Durch die explizite Aufzählung von digitalen Gesundheitsanwendungen im Wortlaut des SGB V soll nun klargestellt werden, dass die gesetzliche Krankenversicherung die Versorgung mit DiGA bei gesundheitlichen Problemen im Zusammenhang mit der Schwangerschaft abdeckt.
Als neuer Wortlaut im SGB V ist nun vorgesehen:
In § 24c Satz 1 Nummer 2 werden die Wörter „und Hilfsmitteln“ durch die Wörter „, Hilfsmitteln und digitalen Gesundheitsanwendungen“ ersetzt.
§ 24e wird wie folgt geändert: In Satz 1 werden die Wörter „und Hilfsmitteln“ durch die Wörter „Hilfsmitteln und digitalen Gesundheitsanwendungen“ ersetzt.
Weitere Änderungen betreffen die Paragrafen zur Zuzahlungsbefreiung bei Schwangerschaft. Eine nähere Erläuterung zur Abgrenzung des Versorgungsanspruchs bei regulär verlaufender Schwangerschaft steht jedoch aus, sodass diese Aufnahme in das Gesetz weiterhin Interpretationsspielraum bietet. Denn eine Verschreibung von DiGA allein aufgrund einer Schwangerschaft ist weiterhin nicht möglich.
2.3 Preisgestaltung anhand von Erfolgsmessungen
Zu beachten ist die obligatorische Einführung einer anwendungsbegleitenden Erfolgsmessung für alle im Verzeichnis gelisteten DiGA zum 1.1.2026. Die Ergebnisse dieser Erfolgsmessung müssen kontinuierlich an das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) gemeldet und im Verzeichnis veröffentlicht werden. Das Bundesgesundheitsministerium sieht im DigiG außerdem einen Anteil von 20 % „erfolgsabhängiger Preisbestandteile” vor.
Denkbare Erfolgskriterien wären laut Überlegungen des BMG zum Beispiel, wie viele Stunden die Patienten eine DiGA durchschnittlich pro Tag nutzen und ob die Anwendung abgebrochen wird, im Gesetz sind jedoch keine konkreten Kriterien genannt. Da jedoch weder der Spitzenverband der gesetzlichen Krankenkassen noch die Hersteller die vom BMG vorgeschlagenen Kriterien für sinnvoll halten, wurde die verpflichtende Einführung der erfolgsabhängigen Preisbestandteile auf den 1.1.2026 festgelegt, um eine Bestimmung praxisrelevanterer Kriterien zu ermöglichen.
Diese neue Anforderung stellt eine zusätzliche Herausforderung für DiGA-Hersteller dar, wenn es darum geht, ihre Produkte gewinnbringend auf dem Markt zu positionieren. Einerseits muss ein entsprechendes Tracking umgesetzt werden, außerdem wird dadurch das Thema Adhärenz noch bedeutsamer.
2.4 Keine 14-tägige Rückgabefrist
Im ursprünglichen Gesetzesentwurf war eine 14-tägige Rückgabefrist vorgesehen: Patienten sollten sich eigenständig gegen die Nutzung einer DiGA in den ersten 14 Tagen entscheiden können. In diesem Zeitraum wäre die Nutzung kostenfrei gewesen. Dieses Verfahren wäre einzigartig im Bereich der durch Ärzte verordneten Therapien gewesen. Nicht zuletzt aufgrund des Einspruchs des SVDGV wurde dieser Absatz wieder aus dem Gesetzesentwurf gestrichen.
2.5 Zusendung von Freischaltcodes innerhalb von 2 Tagen durch die Krankenkassen
In der Vergangenheit mussten Patienten teilweise lange auf die Zusendung der Freischaltcodes warten, die zur Nutzung einer DiGA notwendig sind. Die für die Zustellung verantwortlichen Krankenkassen wurden nun dazu verpflichtet, diese Freischaltcodes innerhalb von zwei Tagen zuzusenden.
2.6 Interoperabilität
Interoperable Informationssysteme sind entscheidend für eine hochwertige Gesundheitsversorgung, aber aufgrund der Fragmentierung im deutschen Gesundheitssystem und der Vielfalt der Informationssysteme besteht die Gefahr von Qualitäts- und Quantitätseinbußen beim Austausch behandlungsrelevanter Daten. Um die Interoperabilitätsziele zu erreichen, wurde im DigiG beschlossen, die Verbindlichkeit von Standards und Profilen zu erhöhen, was zu einer verbesserten Datenverfügbarkeit, höherer Behandlungsqualität und stärkerem Schutz der Gesundheit und informationellen Selbstbestimmung der Versicherten führen soll.
Für DiGA verweist das DigiG auf die Regelung nach § 374a SGB V, die ab Mitte 2024 gilt. Damit soll eine größere Offenheit zwischen Hilfsmitteln/Implantaten und DiGA geschaffen werden, wodurch DiGA in Zukunft Zugang zu mehr Datenquellen erhalten sollen. Gerne können Sie sich dazu in unserem Blogartikel informieren.
2.7 Telemedizinisches Monitoring
Im Digital-Gesetz ist vorgesehen, dass Telemedizin einen festen Platz in der Gesundheitsversorgung einnimmt. Insbesondere sollen Videosprechstunden noch weitreichender genutzt und leichter in Anspruch genommen werden können. In einem ersten Schritt wird die bisherige Begrenzung für Videosprechstunden aufgehoben. Für DiGA-Hersteller könnte es interessant sein, dass umfassende Monitoringkonzepte, auch unter Einbeziehung von DiGA, erstattungsfähig werden sollen. Der TI-Messenger soll außerdem in DiGA eingebunden werden können. Da es sich hier vor allem um den weiteren Ausbau bestehender Strukturen handelt, wird die Zukunft zeigen, ob sich für DiGA-Hersteller Anknüpfungspunkte ergeben.
Quelle: Artikel des Handelsblatts zum Entwurf des DigiG
2.8 Innovationsfonds
Der Innovationsfonds soll Brücken zwischen den Versorgungsbereichen im Gesundheitswesen schaffen, die bisher nur mit Schwierigkeiten interagieren können. Finanziert wird er aus den Mitteln der gesetzlichen Krankenversicherung. Ursprünglich war dieser Fonds bis 2024 befristet. Der Innovationsfonds erfährt eine Verstetigung und Erweiterung, um die Entwicklung innovativer Versorgungsformen und Versorgungsforschung zu fördern. Allerdings eröffnen sich für DiGA-Hersteller aufgrund der geltenden Antragsregeln keine neuen Fördermöglichkeiten. Produktbezogene Studien können nicht gefördert werden, und ein unmittelbares wirtschaftliches Interesse an den Studienergebnissen stellt ein Ausschlusskriterium dar.
Mehr Informationen zur Förderung digitaler Gesundheitslöungen über den Innovationsfonds lesen Sie unserem Fachartikel: Innovationsfonds (2025): Förderung für digitale Gesundheitslösungen
2.9 Strukturierte Behandlungsprogramme bei Diabetes
Das DigiG sieht vor, dass der Gemeinsame Bundesausschuss (G-BA) neue strukturierte Behandlungsprogramme für Versicherte mit Diabetes mellitus Typ I und Typ II einführt, die auf digitalisierten Versorgungsprozessen basieren. Durch die Integration bisher getrennter Datenquellen bei Patienten und Leistungserbringern ermöglicht diese Maßnahme einen Versorgungsprozess, der gezielt digitale Technologien einsetzt und so eine effektivere Therapie gestattet. In Punkt 13 der Änderungsvorschläge zum SGB V werden „digitale Gesundheitsanwendungen“ konkret als Teil dieser digitalisierten Versorgungsprozesse erwähnt. Dies birgt interessante Anknüpfungspunkte für DiGA-Hersteller.
2.10 Schreiben der DiGA in die ePA und Implementierung der GesundheitsID
Zum 30.4.2024 muss zur Implementierung der GesundheitsID sowie das Schreiben der DiGA in die elektronische Patienenakte (ePA) umgesetzt sein. Das DigiG verweist hier auf den § 374a SGB V mit dem bereits die gesetzlichen Voraussetzungen dieser Verpflichtung existieren.
2.11 Einführung und Weiterentwicklung des E-Rezepts
Seit 1.1.2024 müssen Rezepte elektronisch ausgestellt werden. Im DigiG sind jedoch auch weitergehende Ziele festgelegt. So soll es möglich sein, die elektronische Gesundheitskarte über ein NFC-fähiges Smartphone einzulesen und so E-Rezepte auch bei Online-Apotheken einzulösen. Die Spezifikationen befinden sich jedoch derzeit noch in der Abstimmung.
3. Der Weg des DigiG durch die Beschlussgremien
Der DigiG-Referentenentwurf des BMG unter Karl Lauterbach musste bis zur endgültigen Verabschiedung im Bundestag noch verschiedene Hürden nehmen. Die unterschiedlichen Verhandlungsgremien inklusive des Zeitplans sind in der folgenden Grafik dargestellt. Am 2. Februar 2024 wurde das Gesetz final beschlossen, die damit gültige Version finden Sie hier.
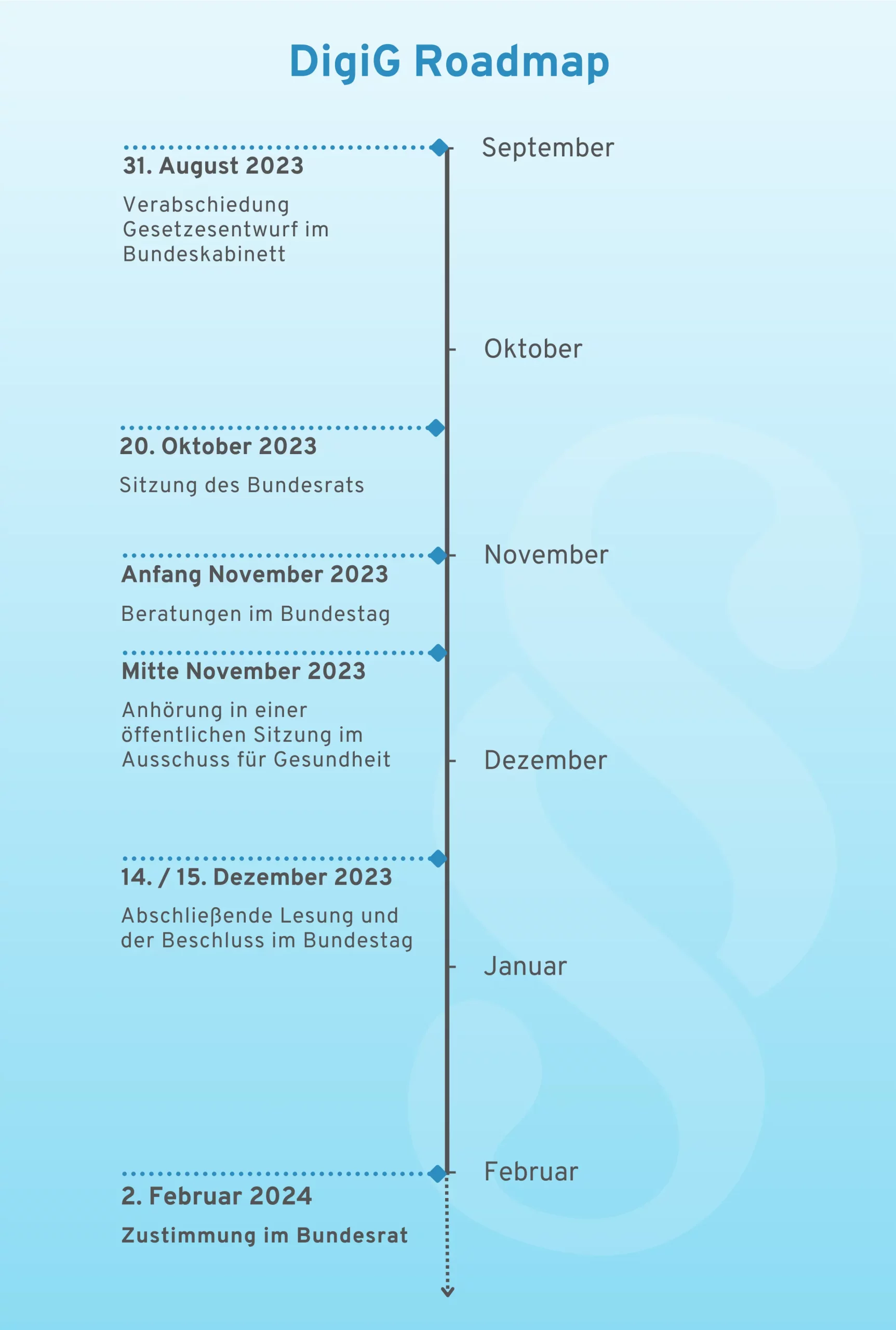
Die DigiG Roadmap
4. Die Stellungnahmen der Interessengruppen zum Entwurf des DigiG
Angesichts der beabsichtigten Einführung des DigiG, hatten sowohl die Krankenversicherungen als auch die Herstellerseite umfassende Stellungnahmen zum Gesetzesentwurf abgegeben. Dadurch kam es zu deutlichen Anpassungen im finalen Gesetz. Im Folgenden erläutern wir die wichtigsten Punkte der damaligen Stellungnahmen:
4.1 Spitzenverband Digitale Gesundheitsversorgung (SVDGV) und Bundesverband Medizintechnologie
Der Spitzenverband Digitale Gesundheitsversorgung (SVDGV) hat 169 Mitglieder. Darunter finden sich auch viele DiGA-Hersteller. Begrüßt wurde die Möglichkeit, zukünftig auch DiGA der Risikoklasse IIb auf den Markt zu bringen. Dass diese DiGA laut DigiG vom Fast-Track-Verfahren ausgeschlossen werden, kann der SVDGV nicht nachvollziehen. Zahlreiche Beispiele aus der Softwarewelt könnten belegen, dass ein Medizinprodukt einer höheren Risikoklasse nicht automatisch komplexer und gefährlicher sei. Der Bundesverband Medizintechnologie sieht durch diese neue Auflage sogar den Erfolg von DiGA in Gefahr. Die Unternehmensvertretungen fordern, dass allen DiGA-Herstellern eine Erprobungsphase zugestanden wird.
Ebenso lehnte der SVDGV den inzwischen gestrichenen Vorschlag im DigiG ab, dass DiGA vom Patienten 14 Tage kostenlos getestet werden können. Die Start-up-Vertretung schlug zudem vor, den Erprobungszeitraum des Fast-Track-Verfahrens zu verlängern. Denn wenn ein Unternehmen erst am Ende der Erprobungsphase Studienergebnisse vorlegen kann, werden diese vom BfArM nicht mehr geprüft. Der SVDGV begrüßte die Pläne, den Anspruch auf DiGA für Schwangerschaft und Mutterschaft explizit im Gesetzesentwurf aufzunehmen. Um deren Gesundheitsversorgung zu verbessern, sollte nach Ansicht des Verbandes jedoch deutlicher werden, dass Schwangere und Mütter immer Anspruch auf eine DiGA haben. Zusätzlich wünschte sich der SVDGV eine Aufnahme normal verlaufender Schwangerschaften über den entsprechenden ICD-10 Code. Die Start-up-Vertretung schlug außerdem vor, künftig auch Verhütungs-Apps als DiGA zuzulassen.
Mehr dazu lesen Sie in der vollständigen Stellungnahme.
4.2 Kassenärztliche Bundesvereinigung
Die Kassenärztliche Bundesvereinigung (KBV) ging in ihrer Stellungnahme zum DigiG nur mit knappen Worten auf DiGA ein. Sie befürwortete, dass die Zulassung von DiGA der Risikoklasse IIb im Gesetzesentwurf mit dem Ausschluss der vorläufigen Zulassung ohne abgeschlossenen Evidenznachweis einhergeht. Zusätzlich plädierte der KBV dafür, dass für die Verordnung von DiGA ein genereller Genehmigungsvorbehalt der Krankenkassen eingeführt wird, wie er auch für andere nicht genehmigungspflichtige Leistungen zur Gesundheit wie zum Beispiel häusliche Krankenpflege gilt.
Lesen Sie die Stellungnahme zum Gesetzesentwurf hier im Volltext.
4.3 GKV-Spitzenverband
Der GKV-Spitzenverband lehnte ebenfalls, wie in Karl Lauterbachs Entwurf zum DigiG vorgesehen, die vorläufige Aufnahme einer App der Risikoklasse IIb ohne Evidenznachweis in das DiGA-Verzeichnis ab. Allerdings hält die Kassenvertretung das sogenannte Fast-Track-Verfahren zur vorläufigen Zulassung vor dem positiven Versorgungsnachweis bereits heute im Ganzen für ungeeignet.
Der GKV-Spitzenverband unterstützte die Nutzung digitaler Gesundheitsanwendungen zur Verbesserung der medizinischen Versorgung von Schwangeren. Die Erweiterung des Anspruchs auf solche Anwendungen betrifft Schwangere mit spezifischen Gesundheitsproblemen, jedoch nicht normale Schwangerschaftsbegleitung oder Präventionsanwendungen. Aufgrund fehlender Klarheit in den bestehenden Gesetzen forderte der GKV-Spitzenverband eine präzisere Definition der Anwendungszwecke und -voraussetzungen durch den Gesetzgeber. Dies solle sicherstellen, dass die Leistungen der gesetzlichen Krankenversicherung gezielt auf die Bedürfnisse von Schwangeren mit Gesundheitsproblemen ausgerichtet sind.
Mehr dazu steht in der Stellungnahme des GKV-Spitzenverbands.
5. Fazit
Das DigiG legt den Kurs für die Zukunft der digitalen Gesundheitsversorgung fest. Insgesamt wird das DigiG begrüßt und als wichtiger Schritt der Digitalisierung im Gesundheitswesen betrachtet. Es ergeben sich aus dem Gesetz jedoch gemischte Auswirkungen für DiGA-Hersteller. Die Möglichkeit der Zulassung von DiGA der Risikoklasse IIb ist positiv zu bewerten. Gleichzeitig stellen der Ausschluss dieser DiGA aus der vorläufigen Zulassung und die verpflichtende Erfolgsmessung Herausforderungen für die Hersteller dar. Auch die weiterhin bestehenden Unklarheiten bezüglich DiGA bei Schwangerschaft könnten sich als Hindernis erweisen. Die genauen Auswirkungen werden sich erst im Verlauf der Zeit zeigen.
The post Das Digital-Gesetz (DigiG): Leitfaden für DiGA-Hersteller appeared first on QuickBird Medical.
]]>The post Validierung von Medizinprodukt-Software nach MDR appeared first on QuickBird Medical.
]]>Doch was genau ist mit der Validierung von Software gemeint? Welche Software muss überhaupt validiert werden? Und wie sieht diese Validierung in der Praxis aus? Unser Leitfaden gibt Ihnen konkrete Antworten auf alle Aspekte dieser Fragen.
Arten der Validierung in Bezug auf Software-Medizinprodukte
Wenn in Bezug auf Software-Medizinprodukte von „Validieren“ die Rede ist, kann sich dies auf völlig unterschiedliche Dinge beziehen. Wir müssen daher erstmal grundlegend verstehen, in welchem Kontext dieser Begriff verwendet wird.
Es gibt im Allgemeinen folgende Kategorien, in denen der Begriff “Software-Validierung” verwendet wird:
- 1. Validierung der eingesetzten Computersoftware: Computer Systems Validation (CSV)
- 2. Validierung des Software-Medizinprodukts selbst
- 2.1 Validierung der Zweckbestimmung Ihres Software-Medizinprodukts: klinische Bewertung
- 2.2 Validierung der Anforderungen aller Stakeholder an das Software-Medizinprodukt: User Needs bzw. Stakeholder Requirement Validierung
- 2.3 Validierung der Gebrauchstauglichkeit Ihres Software-Medizinprodukts nach IEC 62366
Zumeist werden mit „Software-Validierung“ entweder Kategorie #1 oder Kategorie #2a gemeint. Das kommt aber natürlich auf den Kontext an. Daher ist ein holistisches Verständnis dieser Aspekte wichtig. Wir gehen im Folgenden auf alle Kategorien ein und erläutern diese im Detail.
1. Validierung der eingesetzten Computersoftware: Computer Systems Validation (CSV)
Im Kontext der Medizinprodukt-Software-Validierung wird auch häufig von der Validierung der eingesetzten Computer-Software geredet. Ein Hersteller eines MDR-Medizinprodukts muss Computer-Software validieren, die innerhalb des Qualitätsmanagements genutzt wird.
Dies wird in der ISO 13485 4.1.6 weiter spezifiziert: „Die Organisation muss Verfahren für die Validierung der Anwendung der Computersoftware im Qualitätsmanagementsystem dokumentieren.“
Eingesetze Computer-Software bezieht sich hierbei nicht auf Ihr eigentliches Software-Medizinprodukt. Gesprochen wird hierbei von jeglichen eingesetzten Software-Tools, die einen Einfluss auf Ihr Qualitätsmanagementsystem oder die Sicherheit oder Leistungsfähigkeit Ihres Medizinprodukts haben.
Wir sprechen daher gerne auch von „Software-Tools“, weil es den Unterschied zu Ihrem eigentlichen Software-Medizinprodukt und den verwendeten Bibliotheken besser zum Ausdruck bringt. Wichtig: Software-Bibliotheken und Frameworks, die Sie in Ihr Software-Medizinprodukt einbauen, sind keine Software-Tools, sondern SOUP (Software of Unknown Provenance) nach IEC 62304. Diese SOUPs müssen nicht die Validierung der Software-Tools durchlaufen, sondern sind Teil eines gesonderten SOUP-Evaluations-Prozesses, den wir hier nicht näher erläutern. Erfahren Sie mehr zu SOUPs in diesem Artikel: IEC 62304: Software-Lebenszyklus-Prozesse von Medizinprodukten
Beispiele für relevante Software-Tools, die validiert werden müssen, könnten sein:
- Dokumentations-Software: Confluence, Word, Google Docs etc.
- Task-Management-Software: Jira, Todoist etc.
- Entwicklungsumgebungen (IDE) für Ihr Produkt: Xcode, Android Studio, WebStorm, Eclipse etc.
- Dokumenten-Ablage-Software: Dropbox, Google Drive, OneDrive etc.
- usw.
Die Liste Ihrer eingesetzten Tools wird entsprechend sehr lang sein, da für die effiziente Entwicklung einer modernen Software eben sehr viel Fremdsoftware eingesetzt wird.
Das Gute: Sie müssen nicht jedes dieser Software-Tools umfangreich validieren. Der Validierungs-Prozess folgt einem Risiko-basierten Ansatz. Demnach fokussieren Sie sich auf die Software-Tools, die den größten (indirekten) Einfluss auf die Qualität des Produkts und die Patientensicherheit haben. Eine Fehlfunktion in einem Versions-Verwaltungs-System ist demnach verhältnismäßig schwerwiegend, weil ein Anwender so ggf. eine falsche Software-Version ausgeliefert bekommt. Eine Fehlfunktion in einem Bewerbermanagement-System hat hingegen meist kaum Einfluss auf die Produktqualität.
In einem zukünftigen Artikel werden wir genauer beschreiben, wie diese Tools-Validierung abläuft. Melden Sie sich gern hier zum Newsletter an, wenn Sie hierzu benachrichtigt werden möchten:
2. Validierung des Software-Medizinprodukts selbst
2.1 Validierung der Zweckbestimmung Ihres Software-Medizinprodukts: klinische Bewertung
Um Ihr Software-Medizinprodukt (SaMD) in der EU auf den Markt zu bringen, müssen Sie validieren, dass dieses seine medizinische Zweckbestimmung erfüllt. Um dies zu tun, ist unter anderem eine sogenannte klinische Bewertung Ihres Medizinprodukts notwendig. Diese klinische Bewertung verfolgt das Ziel, den klinischen Nutzen Ihres Produkts nachzuweisen, welcher in seiner Zweckbestimmung definiert ist – das versteht man dann unter „Validierung der Zweckbestimmung„. Zur klinischen Bewertung kann zum Beispiel eine Literaturrecherche und die Durchführung eigener Studien zählen.
Doch bevor Sie die Zweckbestimmung Ihres Medizinprodukts validieren können, müssen gewisse Voraussetzungen erfüllt sein, wie im nächsten Kapitel beschrieben wird.
Basis-Arbeiten zur Validierung der Zweckbestimmung
Hierfür müssen Sie im ersten Schritt vor allem erstmal die Zweckbestimmung für Ihr Produkt definieren. Dies ist natürlich die Basis für deren Validierung. Wie genau Sie Ihre Zweckbestimmung Ihres Medizinprodukts finden und definieren, können Sie in unserem umfangreichen Leitfaden nachlesen: Formulierung der Zweckbestimmung für (Software-)Medizinprodukte. Sie sollten selbstverständlich auch überprüfen, ob Ihre Software mit der angegeben Zweckbestimmung überhaupt ein Medizinprodukt nach MDR ist (siehe auch unseren Leitfaden: Ist Ihre App ein Medizinprodukt?). Wenn dies der Fall ist, müssen Sie im nächsten Schritt die Risikoklasse Ihres SaMD bestimmen (siehe auch unseren Leitfaden zur Risiko-Klassifizierung nach MDR), weil diese einen starken Einfluss auf die Gestaltung der Validierung hat.
Klinische Bewertung
Nachdem Sie Ihre Software fertig entwickelt haben, ist es Zeit für die klinische Bewertung bzw. Validierung Ihres Produkts. In diesem Rahmen weisen Sie nach, dass Ihr Produkt sowohl die Zweckbestimmung erfüllt als auch sicher in der Anwendung ist bzw. keine inakzeptablen Gesundheitsrisiken mit sich führt.
Angenommen, Sie haben ein Produkt, das als Zweckbestimmung die Therapie von schweren Depressionen angibt. Hierfür müssten Sie nachweisen, dass die Anwendung/Nutzung Ihrer Software z.B. zur Linderung von Depressions-Symptomen führt. Gleichzeitig müssen Sie nachweisen, dass keine inakzeptablen Gesundheitsrisiken durch die Anwendung entstehen (z.B. der Suizid des Anwenders durch eine falsche Behandlung). Weiterführende Informationen finden Sie in unserem umfangreichen MDR-Guide: Klinische Bewertung von Software-Medizinprodukten
2.2 Validierung der Anforderungen aller Stakeholder an das Software-Medizinprodukt: User Needs bzw. Stakeholder Requirement Validierung
Neben der klinischen Bewertung Ihres Produkts müssen Sie nach IEC 82304 die Anforderungen der relevanten Stakeholder identifizieren und validieren. Stakeholder sind bei einer Patienten-zentrierten Anwendung z.B.:
- Patienten als Hauptnutzer der App
- Ärzte, welche über einen Videochat in der App mit dem Patienten kommunzieren
- Content-Administratoren, die Texte, Bilder und Video einpflegen
- Super-Administratoren, die User Management betreiben
- Gesetzliche Vertreter (z.B. BfArM, die Aufsichtsbehörde oder benannte Stelle)
Die Begriffe Stakeholder-Requirements, User Needs und Nutzer-Anforderungen werden zumeist synonym verwendet. Beispiele für Stakeholder-Requirements teilen sich z.B. in folgende Kategorien:
- Funktionale Anforderungen („Als Nutzer möchte ich Übungen in der App zur Verbesserung meiner Lebensqualität durchführen.“)
- Nicht-Funktionale Anforderungen
- Erfüllung des medizinischen Zwecks der Anwendung
- Patientensicherheit (Gesundheit)
- Cyber-Security-Anforderungen bzw. Anforderungen an die Datensicherheit
- Gesetzliche Anforderungen, wie Labelling (CE-Mark in der App mit Hersteller-Informationen) für MDR-Produkte oder die DSGVO für Datenschutz-Themen
- Nutzerfreundlichkeit der Anwendung
- usw.
Nachdem alle Anforderungen strukturiert identifiziert und beschrieben wurden, müssen diese spätestens nach Fertigstellung der Entwicklung validiert werden. Es muss ein Validierungsplan erstellt werden und nach Abschluss der Validierung ein Validierungs-Bericht. Dies kann für jeden User Need anders aussehen. Einige Beispiele für die Validierung der Stakeholder-Requirements könnten sein:
| Stakeholder-Requirement | Validierungs-Plan | Validierungs-Bericht |
| „Als Nutzer möchte ich Übungen in der App zur Verbesserung meiner Lebensqualität durchführen.“ |
|
|
| „Als Nutzer möchte ich, dass meine Daten sicher gespeichert und verarbeitet werden und somit vor unauthorisiertem Zugriff und Manipulation geschützt sind“ |
|
|
2.3 Validierung der Gebrauchstauglichkeit Ihres Software-Medizinprodukts nach IEC 62366
Für die erfolgreiche Zulassung Ihres Software-Medizinprodukts müssen Sie die Gebrauchstauglichkeit Ihrer Software validieren. Die Norm IEC 62366 definiert konkrete Anforderungen an diese Validierung.
Die Gestaltung der Benutzeroberfläche einer Anwendung sollte demnach nicht nur ansprechend sein, sondern auch dazu beitragen, Fehler bei der Verwendung zu vermeiden. Ein Beispiel wäre eine App zur Steuerung einer Insulinpumpe: Es muss dem Patienten deutlich kommuniziert werden, welcher Knopf eine Insulininjektion auslöst, da eine Überdosis im schlimmsten Fall sogar tödlich sein kann.
Die IEC 62366, eine Norm für die Gebrauchstauglichkeit von Medizinprodukten, beschäftigt sich genau mit diesem Thema. Oft sind Todesfälle bei Medizinprodukten nicht auf technische Fehlfunktionen zurückzuführen, sondern auf mangelhafte Gebrauchstauglichkeit und falsche Anwendung. Das Usability Engineering, also die Gewährleistung der Gebrauchstauglichkeit, ist auch Teil des Risikomanagements. Um sicherzustellen, dass ein Produkt gebrauchstauglich ist, müssen Sie gegebenenfalls im Rahmen der sogenannten summativen Evaluation eine Usability-Studie durchführen.
3. Verifizierung vs. Validierung
Die Unterscheidung zwischen Verifizierung und Validierung ist für viele Hersteller eine Quelle der Verwirrung. Um den Unterschied besser zu verstehen, gibt es eine einfache Faustregel:
- Verifizierung: “Wurde das Produkt richtig umgesetzt?”
- Validierung: “Wurde das richtige Produkt umgesetzt?” oder “Erfüllt das Produkt seinen Zweck und die daran gestellten Anforderungen?”
Die Verifizierung von Software-Medizinprodukt kann zum Beispiel durch Software-System-Tests erfolgen, bei denen man das Ergebnis mit den klaren Produktspezifikationen vergleicht. Beispielsweise könnte man überprüfen, ob eine App Therapieempfehlungen auch so anzeigt, wie es in den Anforderungen beschrieben ist.
Die Validierung hingegen konzentriert sich auf die Wirksamkeit des Produkts und kann bei Medizinprodukten zum Beispiel durch klinische Studien erfolgen. Beispielsweise könnte man überprüfen, ob die von einer App empfohlenen Therapien tatsächlich die Erkrankung lindern.
Fazit zur Validierung von Medizinprodukt-Software
Wenn von Validierung im Kontext von Software als Medizinprodukt gesprochen wird, wird nicht immer das Gleiche gemeint. Die Validierung kommt in verschiedenen Anwendungsbereichen bei der Entwicklung Ihres medizinischen Produkts zum Einsatz.
Besonders wichtig zum Verständnis ist die Unterscheidung zwischen der Validierung von eingesetzter Software und der Validierung des Software-Medizinprodukts selbst.
Bei ersterer kann man als Faustregel sagen, je kritischer eine Software für Ihr Qualitätsmanagementsystem ist, desto strenger muss ihre Validierung ausfallen.
Bei der Validierung des Software-Medizinprodukts selbst spielen die Zweckbestimmung und die Stakeholder Requirements eine zentrale Rolle.
Die obige Übersicht hilft Ihnen hoffentlich besser zu verstehen, welche Aspekte für das Validieren von Software relevant sind und wie diese ablaufen.
Falls Sie eine Medizinprodukt-Software planen, treten Sie gern mit uns in Kontakt (Zum Formular). Wir sind auf die Entwicklung derartiger Produkt spezialisiert und setzen Ihre Software regulatorisch konform um.
The post Validierung von Medizinprodukt-Software nach MDR appeared first on QuickBird Medical.
]]>The post Guide zum §374a SGB V – Interoperabilität für DiGA, Hilfsmittel & Implantate appeared first on QuickBird Medical.
]]>Ab Mitte 2024 gilt eine neue gesetzliche Anforderung, welche die Interoperabilität zwischen Medizingeräten und den vom BfArM zugelassenen Digitalen Gesundheitsanwendungen (DiGA) verbessern soll: der §374a SGB V
Konkret geht es dabei um die Implementierung einer interoperablen Backend-zu-Backend-Schnittstelle. Was das genau bedeutet und was Sie darüber wissen müssen, erfahren Sie in diesem Artikel.
Inhaltsverzeichnis
- 1. Wenig Zeit? Das Wichtigste in Kürze
- 2. Der §374a SGB V im Überblick
- 3. Was Hilfsmittel-/Implantat-Hersteller jetzt machen müssen
- 4. Was DiGA-Hersteller machen müssen
- 5. Fazit: Visionen und Limitationen
1. Wenig Zeit? Das Wichtigste in Kürze
- Der §374a SGB V soll ab 2024 dafür sorgen, dass Daten aus Backends von Hilfsmitteln und Implantaten in einem interoperablen Format an eine Digitale Gesundheitsanwendung (DiGA) übertragen werden können. Dies gilt für all jene Produkte, die von den gesetzlichen Krankenkassen erstattet werden.
- Mit dem Gesetz soll eine größere Offenheit zwischen Hilfsmitteln/Implantaten und DiGA geschaffen werden, wodurch DiGA in Zukunft Zugang zu mehr Datenquellen erhalten sollen.
- Um diesen interoperablen Datentransfer umzusetzen, wird aktuell eine Festlegung der KBV entwickelt – das DiGA Device Toolkit.
- Das neue Gesetz könnte bisher unbekanntes Potenzial von DiGA ausschöpfen und das Design zukünftiger Anwendungen maßgeblich beeinflussen. Durch den Zugang zu verschiedenen Daten aus Hilfsmitteln und Implantaten unterschiedlicher Hersteller können diese auch im Sinne der NutzerInnen genutzt werden.
- Insgesamt gibt es aber noch einige Faktoren, die diese Visionen bremsen – zum Beispiel die Tatsache, dass aktuell nur wenige Hilfsmittel/Implantate Daten an ein Backend senden und die Beschränkung auf Hilfsmittel und Implantate, die von den gesetzlichen Krankenkassen erstattet werden. Für viele Produkte gibt es nämlich meist keine Erstattung (z.B. Gewichtswaagen, Fitnesstracker, etc.).
2. Der §374a SGB V im Überblick
(1) Hilfsmittel oder Implantate, die zu Lasten der gesetzlichen Krankenversicherung an Versicherte abgegeben werden und die Daten über den Versicherten elektronisch über öffentlich zugängliche Netze an den Hersteller oder Dritte übertragen, müssen ab dem 1. Juli 2024 ermöglichen, dass die von dem Hilfsmittel oder dem Implantat verarbeiteten Daten auf der Grundlage einer Einwilligung des Versicherten in geeigneten interoperablen Formaten in eine in das Verzeichnis nach § 139e Absatz 1 aufgenommene digitale Gesundheitsanwendung übermittelt und dort weiterverarbeitet werden können, soweit die Daten von der digitalen Gesundheitsanwendung zum bestimmungsgemäßen Gebrauch durch denselben Versicherten benötigt werden.
Hierzu müssen die Hersteller der Hilfsmittel und Implantate nach Satz 1 interoperable Schnittstellen anbieten und diese für die digitalen Gesundheitsanwendungen, die in das Verzeichnis nach § 139e aufgenommen sind, öffnen. Die Beeinflussung des Hilfsmittels oder des Implantats durch die digitale Gesundheitsanwendung ist unzulässig und technisch auszuschließen.
Als interoperable Formate gemäß Satz 1 gelten in nachfolgender Reihenfolge:
- Festlegungen für Inhalte der elektronischen Patientenakte nach § 355,
- empfohlene Standards und Profile im Interoperabilitätsverzeichnis nach § 385,
- offene international anerkannte Standards oder
- offengelegte Profile über offene international anerkannte Standards, deren Aufnahme in das Interoperabilitätsverzeichnis nach § 385 beantragt wurde.
[…]
2.1 Warum das Ganze?
Der angestrebte Nutzen des §374a SGB V ist die Steigerung der Effizienz von DiGA durch den Zugang zu mehr Datenquellen. Insgesamt soll eine größere Offenheit am Markt entstehen, da durch das Gesetz Produkte unterschiedlicher Hersteller in Kombination mit einer DiGA verwendet werden können. Aktuell ist die Offenheit zwischen DiGA und Hilfsmitteln/Implantaten nämlich begrenzt.
Zwar müssen DiGA-Hersteller interoperable Schnittstellen zu externen Medizingeräten anbieten und die verwendeten Standards und Profile offenlegen, allerdings gibt es keine Verpflichtung vonseiten der Hilfsmittel- und Implantat-Hersteller, diese Schnittstellen auch zu nutzen.
Zudem beziehen sich diese Schnittstellen in den meisten Fällen auf Offline-Verbindungen, wie Bluetooth. Interoperabilität zwischen unterschiedlichen Backends, in denen Daten gespeichert sind, ist aktuell nur selten gegeben.
Das Ziel des §374a SGB V lässt sich sehr gut am Beispiel Diabetes erklären:
Immer häufiger werden zu Diabetes-Hilfsmitteln auch Apps entwickelt, welche entsprechende Daten für Betroffene speichern und visualisieren. Das führt aktuell dazu, dass viele Hersteller ihre eigenen Apps entwickeln, die nur mit den eigenen Geräten kompatibel sind.
Stellen wir uns also vor, eine Patientin nutzt ein Glukosemessgerät mit einer dazugehörigen App als digitales Diabetes-Tagebuch. Die Patientin verfügt ebenso über eine Insulinpumpe eines anderen Herstellers, welche ebenfalls Daten an eine App überträgt.
In diesem Fall ist die Patientin dazu gezwungen, zwei unterschiedliche Apps zu verwenden, die nicht sinnvoll miteinander interagieren können, da ein Datenimport in die jeweils andere App nicht unterstützt wird. Beide Apps speichern die Daten aber in ihrem eigenen Backend – wäre es daher nicht sinnvoll, diese Daten zusammenzuführen? Genau hier soll der §374a SGB V in Zukunft eine Verbesserung erreichen.
Dies könnte auch dazu führen, dass DiGA-Hersteller auf bereits verfügbare Geräte zurückgreifen können und keine eigenen (mit der DiGA kompatiblen) Produkte anbieten müssen.
Der Hersteller des Geräts soll nämlich dazu verpflichtet werden, eine Datenübertragung in DiGA anzubieten, wodurch Daten von Geräten verschiedener Hersteller in einer einzigen App verarbeitet werden können.
2.2 Für wen gilt das neue Gesetz?
Ob die Vorgaben des §374a SGB V für Sie als Hilfsmittel- oder Implantat-Hersteller gelten, lässt sich relativ schnell herausfinden. Wie im Gesetzestext erwähnt, ist dies der Fall, wenn die folgenden Kriterien auf Sie zutreffen:
- Die Kosten für Ihr Produkt werden von den gesetzlichen Krankenkassen übernommen.
- Ihr Produkt sendet Daten über den User an Sie (den Hersteller) oder Dritte über öffentlich zugängliche Netze (z.B. Internet).
Wichtig: eine Bluetooth-Verbindung beispielsweise ist hier laut DVPMG nicht gemeint. Im Allgemeinen betrifft die Regelung “[…]Hilfsmittel und Implantate, die elektronisch Daten übermitteln und dem Hersteller oder Dritten über das Internet zur Verfügung stehen […]”.
Die Vorgaben müssen von betroffenen Herstellern bis spätestens 01.07.2024 umgesetzt werden. Eine Ausnahme gibt es für Hilfsmittel und Implantate, die aus medizinischen Gründen notwendig sind und für die es keine Alternative gibt – denn ein Schaden an PatientInnen soll natürlich verhindert werden.
2.3 Was ist eine Digitale Gesundheitsanwendung?
Eine Digitale Gesundheitsanwendung (DiGA), oder “App auf Rezept” ist eine mobile- oder Web-Anwendung, die als Medizinprodukt niedriger Risikoklasse qualifiziert ist und vom BfArM zugelassen wurde. Beispiele dafür könnten zum Beispiel Diabetes-Tagebücher oder digitale Psychotherapie-Kurse sein.
Für eine genauere Definition von DiGA, lesen Sie unseren Artikel zu diesem Thema: Ist Ihre App eine DiGA? Definition & Kriterien digitaler Gesundheitsanwendungen
2.4 Was heißt Interoperabilität?
Sie können mit einem Briefumschlag nicht telefonieren und auch keinen sinnvollen Text auf Spanisch an Ihre englischsprachige Freundin schicken. Interoperabilität beschreibt im Allgemeinen die Fähigkeit von Systemen, zusammenzuarbeiten.
Dazu ist es natürlich notwendig, sich vorab auf gemeinsame Kommunikationskanäle und eine Sprache zu einigen. Das Ziel des §374a SGB V ist grob gesagt, dass alle betroffenen Hilfsmittel und Implantate dieselbe Sprache mit den DiGA sprechen.
So soll in Zukunft erreicht werden, dass sowohl die Daten eines Backends A, als auch jene eines Backends B in eine DiGA übertragen und weiterverarbeitet werden können. Lesen Sie mehr zum Thema Interoperabilität in unserem detaillierten Artikel darüber: Interoperabilität in der Medizin: An einfachen Beispielen erklärt
An dieser Stelle ist es wichtig, noch einmal zu betonen, dass es um die Schnittstelle zwischen dem Backend des Hilfmittels/Implantats und dem Backend der DiGA geht – also wenn Sie Daten über den User in irgendeiner Form über das Internet übertragen.
Datenübertragungen über beispielsweise Kabel oder Bluetooth sind vom §374a SGB V nicht betroffen. Um eine DiGA über eine solche Schnittstelle anzusteuern, ist die ISO/IEEE 11073 Ihre erste Anlaufstelle. DiGA-Hersteller sind dazu verpflichtet, auch abseits vom Backend eine offene Schnittstelle anzubieten, sofern diese Daten aus externen Geräten einlesen. Mehr dazu lesen Sie hier: Interoperabilität für digitale Gesundheitsanwendungen (DiGA)
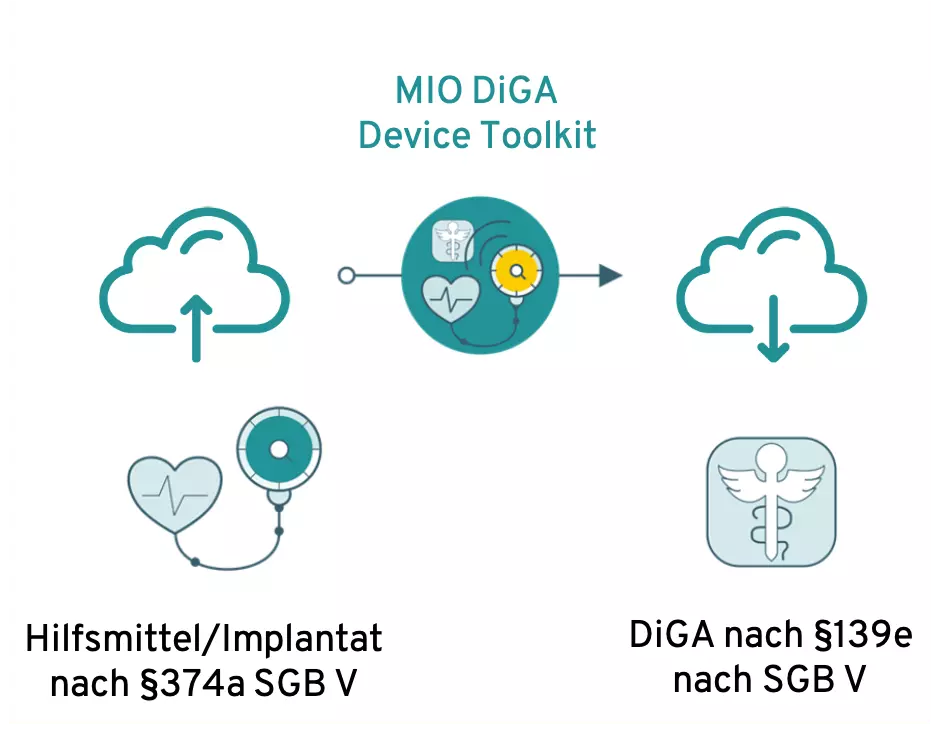
Das MIO DiGA Device Toolkit soll als Schnittelle fungieren
Zur Umsetzung der interoperablen Schnittstelle zwischen Backends von DiGA und Hilfsmitteln/Implantaten ist derzeit ein medizinisches Informationsobjekt (MIO) geplant: Das DiGA Device Toolkit.
Dabei handelt es sich um eine Festlegung der KBV, welche speziell im ePA- und DiGA-Bereich für standardisierte Schnittstellen sorgen sollen.
3. Was Hilfsmittel-/Implantat-Hersteller jetzt machen müssen
Was Sie heute bereits machen können, ist zu evaluieren, ob Sie Daten Ihrer NutzerInnen in einem Backend speichern oder diese an Dritte übertragen.
Wenn das der Fall ist, sollten Sie sich frühzeitig mit der Umsetzung der interoperablen Schnittstelle befassen, da diese bestimmt einige Zeit in Anspruch nehmen wird. Auch wenn das DiGA Device Toolkit am heutigen Tag noch nicht festgelegt ist (21.04.2022), kann die Implementierung bereits geplant und die Auswirkungen evaluiert werden.
Theoretisch müssen nicht alle Daten aus dem Hilfsmittel/Implantat in das Backend der DiGA übertragbar sein – denn das Gesetz beschränkt sich hier auf jene Daten, die von der DiGA “für den bestimmungsgemäßen Gebrauch” benötigt werden.
Hier ist allerdings Vorsicht geboten, denn Sie haben keinen Einfluss darauf, welche Daten von zukünftigen DiGA “gebraucht” werden, weshalb Sie bestimmt auf der sicheren Seite sind, wenn Sie die interoperable Datenübertragung für alle Daten anbieten.
Sprechen Sie uns gerne an, wenn Sie weitere Expertise in diesem Bereich oder Unterstützung bei der Implementierung benötigen.
3.1 Was Sie nicht machen müssen
Wenn Sie aktuell keine Daten über ein Backend speichern oder übertragen, kann das auch in Zukunft so bleiben. Sie müssen in diesem Fall nicht extra Ihr System erweitern, um einen interoperablen Datenaustausch mit DiGA zu ermöglichen.
4. Was DiGA-Hersteller machen müssen
Für DiGA-Hersteller bringt der §374a SGB V keine weiteren Verpflichtungen mit sich. Gerade für DiGA, die keine Daten von externen Geräten empfangen, ist die Regelung zu vernachlässigen.
Allerdings eröffnen sich in Zukunft neue Möglichkeiten für alle Anwendungen, die Daten aus externen Geräten für Ihren bestimmungsgemäßen Gebrauch benötigen. Denn diese Daten könnten durch das Gesetz schon bald einfacher zugänglich sein.
Die Idee ist, dass Sie für Ihre Anwendung keine eigenen Hardware-Geräte mehr anbieten müssen, da die App mit vergleichbaren Geräten kompatibel ist, die bereits von anderen Herstellern auf dem Markt verfügbar sind.
Die Interoperabilitäts-Anforderungen für DiGA finden Sie in diesem Artikel: Interoperabilität für digitale Gesundheitsanwendungen (DiGA)
5. Fazit: Visionen und Limitationen
Die Idee, möglichst viele Gesundheitsdaten an einem Ort zu verarbeiten und miteinander in Beziehung setzen zu können, gibt es schon seit vielen Jahren. Bei der Vorstellung, dass etwa Diabetes-Apps in Zukunft nicht nur Glukose-Messwerte, sondern auch auf Daten aus Körpergewichtswaagen, Fitness-Trackern, CGM-Sensoren oder Insulinpumpen zugreifen können, sind der Fantasie des Möglichen kaum Grenzen gesetzt.
Dadurch sind bisher unbekannte Verbesserungen in der Gesundheitsversorgung möglich, denn je mehr Daten(quellen) einer DiGA zur Verfügung stehen, desto effizienter können diese ihre NutzerInnen unterstützen.
Beispielsweise können Anzeichen für eine Verschlechterung des Gesundheitszustands früh erkannt und rechtzeitig Handlungsempfehlungen gegeben werden.
Zumindest in der Theorie. Denn ein Gesundheitsmarkt, in dem alle Hersteller zusammenarbeiten und ihre Systeme offen für andere gestalten, liegt noch (weit) in der Zukunft.
Inwieweit man Visionen dieser Art nachgehen darf, bleibt nämlich abzuwarten, da der §374a SGB V nur ein vergleichsweise schlankes Anwendungsgebiet hat. Viele Geräte, die von PatientInnen verwendet werden, werden heute nämlich gar nicht, oder nur selten von den gesetzlichen Krankenkassen erstattet und sind vom Gesetz daher nicht betroffen.
Außerdem kritisiert BVMed, dass Hersteller oft nicht wüssten, welche Geräte von wem bezahlt wurden, da ihnen Informationen zu den PatientInnen fehlen würden. Theoretisch muss die interoperable Schnittstelle nämlich nur verfügbar sein, sobald eine gesetzliche Krankenkasse für die Kosten aufgekommen ist. Ein entsprechender Nachweis sei aber unpraktikabel. Außerdem habe man sich eine etwas längere Umsetzungsfrist gewünscht.
Eine weitere Limitation des §374a SGB V ist, dass aktuell nur wenige Geräte ihre Daten an ein Backend des Herstellers oder einer Drittpartei übertragen – vielmehr finden Datentransfers nicht online, sondern beispielsweise über Bluetooth statt.
Erfahren Sie mehr über Interoperabilität in der Medizin im Allgemeinen oder die spezifischen Anforderungen für DiGA.
The post Guide zum §374a SGB V – Interoperabilität für DiGA, Hilfsmittel & Implantate appeared first on QuickBird Medical.
]]>The post Zulassung & Zertifizierung von Software-Medizinprodukten (MDR) appeared first on QuickBird Medical.
]]>Inhalt
- 1. Vorbereitung der Zertifizierung
- 2. Zulassung der Software
- 3. Rahmenbedingungen für die Zulassung von Software-Medizinprodukten
- 4. Fazit

Zeitplan für die Zulassung von Medizinprodukten
1. Vorbereitung der Zertifizierung
Die Vorbereitung auf die Zertifizierung beginnt in jedem Fall noch vor der eigentlichen Entwicklung Ihrer Medizinprodukt-Software. Denn der Aufwand, der für eine reibungslose Zulassung betrieben werden muss, ist stark von der Risikoklasse abhängig, welche Sie in den ersten Schritten des Projekts definieren. Diese entscheidet nämlich über das anzuwendende Konformitätsbewertungsverfahren und die Notwendigkeit einer benannten Stelle für die Zulassung. Was aber in allen Fällen notwendig ist, ist der Aufbau eines Qualitätsmanagementsystem. Dieses soll die Qualität Ihres Produkts sicherstellen und ist die Grundlage für die technische Dokumentation, welche für Medizinprodukte aller Risikoklassen erstellt werden muss.
1.1 Qualifizierung und Klassifizierung
Ganz zu Beginn einer jeden medizinischen Software steht die Definition des angestrebten Nutzens – genauer, die “Zweckbestimmung”. Denn diese entscheidet fundamental darüber, ob es sich bei dem geplanten Produkt überhaupt um ein Medizinprodukt handelt (Qualifizierung). Im darauffolgenden Schritt gilt es, das Produkt entsprechend zu klassifizieren und einer Risikoklasse (I, IIa, IIb oder III) zuzuordnen. Der vorliegende Artikel konzentriert sich auf die Zulassung und Zertifizierung Ihres Software-Medizinprodukts. Ein detaillierter Exkurs in die Qualifizierung und Klassifizierung von Software würde hier den Rahmen sprengen. Selbstverständlich haben wir zu diesen Themen eigene Artikel verfasst, welche Sie hier finden:
- Formulierung der Zweckbestimmung für (Software-)Medizinprodukte
- Leitfaden: Ist Ihre App ein Medizinprodukt?
- Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden
Wichtig ist, dass Sie die Klassifizierung Ihrer Software gewissenhaft durchführen und sich frühzeitig mit diesem Thema beschäftigen. Denn von der Risikoklasse hängt unter anderem das anzuwendende Konformitätsbewertungsverfahren ab, welches weiter unten in diesem Artikel thematisiert wird und den Aufwand, die Dauer und Kosten für das Projekt maßgeblich beeinflusst. Falls Sie noch kein Qualitätsmanagementsystem in Ihrem Unternehmen etabliert haben, gilt es im nächsten Schritt, ein solches aufzubauen (unabhängig von der Risikoklasse). Was Sie grundsätzlich bei der Medizinproduktentwicklung beachten müssen, erfahren Sie in diesem Artikel: Leitfaden für die Entwicklung von Medical Apps: Darauf müssen Hersteller achten
1.2 Auswahl eines Konformitätsbewertungsverfahrens
„Konformitätsbewertung“ bezeichnet das Verfahren, nach dem festgestellt wird, ob die Anforderungen dieser Verordnung an ein Produkt erfüllt worden sind; (Zitat aus Artikel 2 MDR)
Damit Ihre Software eine CE-Kennzeichnung tragen darf, müssen Hersteller zuerst ein sogenanntes Konformitätsbewertungsverfahren durchführen. Die MDR bietet von Anhang IX bis XI drei verschiedene Bewertungsverfahren. Die Auswahl des anzuwendenden Verfahrens ist ebenfalls noch in der Planungsphase des Projekts zu treffen. An dieser Stelle sagen wir gleich, dass für Software eigentlich nur das Verfahren in Anhang IX praktikabel ist: Konformitätsbewertung auf der Grundlage eines Qualitätsmanagementsystems und einer Bewertung der technischen Dokumentation. Zwar ist es auch möglich, ein Bewertungsverfahren anhand einer sogenannten “Baumusterprüfung” durchzuführen, was aber in den meisten Fällen nicht sinnvoll ist. In einem Dokument (NB-MED/2.2/Rec4), das die Position der benannten Stellen zu diesem Thema angibt, steht wörtlich:
“[…] a pure product related evaluation without consideration of the design process is not considered adequate. Consequently, the use of some of the Conformity Assessment Procedures (CAP), as defined by the directives may be unsuitable for software.”
Ferner beschreibt das Dokument die Notwendigkeit von Lebenszyklusprozessen. Dies würde im Grunde bedeuten, dass die Produktentwicklung den Vorgaben der IEC 62304 folgen muss, welche ebenfalls ein Qualitätsmanagementsystem erfordert. Von daher stellt sich ohnehin die Frage, warum Hersteller dann nicht gleich das Konformitätsbewertungsverfahren nach Anhang IX wählen. Die Anwendung des Konformitätsbewertungsverfahrens in Anhang IX (und der Einbezug einer benannten Stelle) ist für Hersteller von Medizinprodukten der Klasse I nicht erforderlich. Das heißt aber nicht, dass Sie für ein Klasse I Produkt nichts machen müssen – ein Qualitätsmanagementsystem und die technische Dokumentation für Ihr Produkt benötigen Sie dennoch. Dies wird aber nicht von einer benannten Stelle überprüft. Doch welche Art von Software fällt überhaupt noch in Klasse I? Lesen Sie mehr dazu in diesem Artikel: Klassifizierung von Software-Medizinprodukten: MDR Leitfaden Neben dem Konformitätsbewertungsverfahren müssen Sie als Hersteller auch eine EU-Konformitätserklärung für Ihr Produkt unterzeichnen.
Info: Unterschied zwischen Qualitätsmanagmentsystem und technischer Dokumentation:
- Ein Qualitätsmanagementsystem beschreibt die Prozesslandschaft in Ihrem Unternehmen und ist produktunabhängig. Prozesse sind Arbeitsabläufe, die durch bestimmte Trigger angestoßen und durchlaufen werden und erzeugen immer einen oder mehrere Outcomes. Risikomanagement wäre ein klassisches Beispiel für einen Prozess (welcher natürlich auch in mehrere Teilprozesse untergliedert werden kann).
- Die technische Dokumentation ist hingegen produktspezifisch und sie enthält alle Dokumente und Aufzeichnungen, die das Produkt betreffen. Darunter fallen beispielsweise Pläne und Reports (z.B. für das Risikomanagement oder die klinische Bewertung).

Unterschied zwischen Qualitätsmanagementsystem und technischer Dokumentation
1.3 Aufbau eines Qualitätsmanagementsystems
Bei Ihrem geplanten Produkt handelt es sich um ein Medizinprodukt? Glückwunsch, denn dann werden Sie bald auch ein eigenes Qualitätsmanagementsystem haben! Ein solches ist nämlich verpflichtend für alle Software-Medizinprodukthersteller und legt die Grundlage für den Großteil der technischen Dokumentation, die Sie gemäß MDR für Ihr Produkt erstellen müssen. Während die MDR keine wirklich gute Anleitung für den Aufbau eines QMS ist, halten Sie sich zunächst am besten an die ISO 13485 – das ist auch keine gute Anleitung, aber zumindest in Bezug auf die Anforderungen an vielen Stellen konkreter. Sie bekommen aber keinen Schritt-für-Schritt-Guide, der Ihnen sagt, was genau Sie machen müssen. Daher werden Sie sehr wahrscheinlich externe Unterstützung in Form von Beratungsdienstleistungen benötigen. Der Aufbau eines QMS nimmt je nach Unternehmensgröße und Komplexität bestehender Strukturen erfahrungsgemäß einige Monate in Anspruch. Achtung: Insgesamt sind die Anforderungen der MDR an Qualitätsmanagementsysteme etwas umfangreicher, als jene der ISO 13485. Die Norm legt aber eine gute Basis und deckt die meisten Anforderungen der MDR ab. Welche weiteren Normen Sie bei der Entwicklung von Software-Medizinprodukten berücksichtigen sollten, erfahren Sie in diesem Artikel: Leitfaden für die Entwicklung von Medical Apps: Darauf müssen Hersteller achten.
1.4 Erstellung der technischen Dokumentation
Die technische Dokumentation ist in den Anhängen II und III der MDR spezifiziert. Sie soll Ihnen als Hersteller in erster Linie dabei helfen, bessere Produkte im Sinne der Leistungsfähigkeit und Patientensicherheit zu entwickeln und beinhaltet zum Beispiel Planungsdokumente, Berichte und Aufzeichnungen, die während des Produkt-Lebenszyklus entstehen. Zu berücksichtigen sind hier auch die Beschreibung geplanter Aktivitäten nach der Inverkehrbringung. Ein großer Teil der technischen Dokumentation geht aus den Prozessen Ihres Qualitätsmanagementsystems hervor, was bedeutet, dass vieles davon automatisch entsteht, wenn Sie sich an Ihre Prozesse halten. Die technische Dokumentation ist grob gesagt eine Sammlung aller Informationen, die das Produkt betreffen.
1.4.1 Durchführung einer klinischen Prüfung
In vielen Fällen müssen Sie für Ihr Produkt auch eine klinische Prüfung durchführen. Diese ist ebenfalls ein Teil der technischen Dokumentation und fällt in den Bereich der klinischen Bewertung. Mehr über die klinische Bewertung lesen Sie in diesem Artikel: MDR-Guide: Klinische Bewertung von Software-Medizinprodukten.
1.5 Suche nach einer benannten Stelle (für Klasse IIa oder höher)

Wann eine benannte Stelle notwendig ist
Info: Es kann auch seltene Ausnahmefälle geben, in denen auch für Medizinprodukte der Klasse I eine benannte Stelle erforderlich ist (z.B. wiederverwendbare chirurgische Instrumente). Das trifft auf Software aber in den allermeisten Fällen nicht zu. Produkte der Klasse IIa oder höher benötigen das “Go” einer benannten Stelle, bevor sie mit einer CE-Kennzeichnung auf den Markt gebracht werden können. Und spätestens hier kann die Planung der Medizinprodukt-Zertifizierung stark ins Schleudern geraten. Eine verspätete Suche nach einer benannten Stelle kann die Zulassung Ihrer Software nämlich um mehrere Monate verzögern – ein Szenario, das Sie als Medizinprodukt-Hersteller natürlich vermeiden möchten. Insgesamt gibt es 2 Hauptgründe, weshalb wir Ihnen empfehlen, sich frühzeitig auf die Suche zu begeben:
- Die Nachfrage ist größer als das Angebot: Wartezeiten von mehreren Monaten sind durchaus üblich. Konkret gibt es in Deutschland im Moment nur 7 benannte Stellen, die eine Prüfung nach MDR durchführen können (Stand März, 2022). Alle benannten Stellen nach MDR finden Sie im Nando-Verzeichnis.
- Nicht alle benannten Stellen haben eine gleich große Software-Affinität. Mit anderen Worten: manche Stellen machen Ihnen den Prozess unnötig schwer, da sie nicht verstehen, dass sich Software-Entwicklung von klassischer Medizinprodukt-Produktion in vielen Punkten unterscheidet. Es erspart Ihnen einiges an Zeit, und vor allem Nerven, wenn Sie einen Auditor erwischen, der mit Software-Produkten vertraut ist und “Ihre Sprache spricht”. Zudem verstehen Software-affine Stellen auch besser, welche Maßnahmen in einem QMS sinnvoll sind und die Chancen sind geringer, dass sie sich im Audit auf Prozesse fokussieren, die für Software wenig relevant sind.
Die Suche nach einer geeigneten benannten Stellen beginnt also in der Regel im oben verlinkten Nando-Verzeichnis. Wir möchten an dieser Stelle keine Empfehlung für oder gegen eine der benannten Stellen abgeben, aber folgende Quellen könnten bei der Auswahl einer benannten Stelle hilfreich sein:
- Wenn möglich, holen Sie sich Erfahrungsberichte von anderen Software-Medizinprdoukt-Herstellern ein
- Fragen Sie explizit nach Erfahrungen mit Software-Medizinprodukten in ersten Beratungsgesprächen mit den benannten Stellen
- Sie haben bereits ein ISO 13485 Zertifikat? Sehen Sie nach, ob Ihre Zertifizierungsstelle auch als benannte Stelle zertifiziert ist (vorausgesetzt, Sie sind mit dieser zufrieden)
- Sprechen Sie uns an
Falls Sie auch eine Zertifizierung nach ISO 13485 anstreben, macht es Sinn, diese bei einer Organisation zu beantragen, die auch als benannte Stelle zertifiziert ist – so bekommen Sie beides aus einer Hand, müssen Ihre Zertifizierungsstellen nicht dauernd wechseln und werden nicht Opfer von unterschiedlichen Interpretationen der einzelnen Organisationen.
2. Zulassung der Software
Sie haben ein Qualitätsmanagementsystem etabliert und Ihre technische Dokumentation erstellt? Super! In diesem Kapitel erfahren Sie, worauf Sie alles achten müssen, bevor Sie Ihr Produkt auf den Markt bringen können.
2.1 Prüfung durch die benannte Stelle (für Klasse IIa oder höher)
Wie schon weiter oben beschrieben, muss bei Produkten der Klasse IIa oder höher eine benannte Stelle für die Zulassung Ihres Medizinprodukts mit einbezogen werden. Dabei werden zwei Dinge (üblicherweise in dieser Reihenfolge) auditiert:
- Die technische Dokumentation
- Ihr Qualitätsmanagementsystem
Die Prüfung der technischen Dokumentation kann mehrere Feedbackschleifen beinhalten und sich je nach Bedarf auch über mehrere Wochen und Monate ziehen. Sobald die technische Dokumentation abgesegnet wurde, wird Ihr Qualitätsmanagementsystem auditiert (diese Reihenfolge ist üblich, aber nicht verpflichtend). Als Faustregel gilt: Je größer Ihr Unternehmen, desto länger dauert der Audit der benannten Stelle vor Ort. Stellen Sie sich also auf jeden Fall auf mehrere Tage ein, in denen die Auditoren zu Besuch kommen (in Ausnahmefällen wird der Audit auch Remote durchgeführt)).
Sie verfügen über ein ISO 13485 Zertifikat?
Rein formell macht eine ISO 13485-Zertifizierung keinen Unterschied – dadurch wird der Audit Ihres Qualitätsmanagementsystems durch die benannte Stelle weder verkürzt, noch fällt er ganz weg. Das liegt daran, dass der Scope der ISO 13485 nicht ganz deckungsgleich mit jenem der MDR ist, wenn es um Qualitätsmanagementsysteme geht. Zudem können Sie eine ISO Zertifizierung auch von Organisationen erhalten, die nicht als benannte Stelle zertifiziert sind. Dennoch ist ein ISO 13485 Zertifikat natürlich von Vorteil, weil Ihr Auditor mit einem ganz anderen Gefühl auf Sie zukommt, wenn ein solches vorhanden ist. Das signalisiert nämlich, dass Ihr Qualitätsmanagementsystem im Großen und Ganzen “schon in Ordnung” sein muss und die Wahrscheinlichkeit für grobe Abweichungen geringer ist. Zudem kommen Sie weniger in Erklärungsnot bei der Frage, warum sie glauben, dass Ihr Qualitätsmanagementsystem MDR-konform ist. Tipp: Falls Sie noch kein ISO 13485 Zertifikat für Ihr Qualitätsmanagementsystem haben, empfiehlt es sich, dieses im Zuge des QMS-Audits zu beantragen. Fragen Sie bei Ihrer benannten Stelle nach, ob das möglich ist. Das spart Ihnen möglicherweise einen zweiten Audit. Rechtlich gesehen benötigen Sie aber kein ISO 13485 Zertifikat.
2.1.1 Sonderfall bei Risikoklasse I
Wie bereits oben erwähnt, benötigen Hersteller von Klasse I Produkten keine benannte Stelle für die Konformitätsbewertung ihrer Software. Es reicht in diesem Fall, ein Qualitätsmanagementsystem zu etablieren, die technische Dokumentation nach Anhang II und III anzufertigen und im Anschluss die EU-Konformitätserklärung zu erstellen. Danach kann die Software als Medizinprodukt im DMIDS bzw. EUDAMED angemeldet werden (mehr dazu weiter unten) und Sie haben Ihre Pflichten als Hersteller erfüllt. Somit kann das Produkt guten Gewissens mit der CE-Kennzeichnung auf den Markt gebracht werden. Aber gibt es gar keine Prüfung von der Aufsichtsbehörde? Nicht zwangsläufig – zumindest ist der Markteintritt nicht davon abhängig. Offiziell sind Behörden nämlich nicht dazu verpflichtet, derartige Produkte direkt bei der Registrierung im Detail zu prüfen. Aber gerade bei der ersten Anmeldung eines Produkts ist es durchaus realistisch, dass die technische Dokumentation und/oder Ihr QMS zumindest stichprobenartig angeschaut wird. Stellen Sie sich also darauf ein, dass Sie einige Zeit nach der Anmeldung des Produkts von Ihrer Aufsichtsbehörde aufgefordert werden, ihr Einsicht in ihre technische Dokumentation zu gewähren (z.B. in das Risikomanagement oder die klinische Bewertung). Auf eine solche Anfrage müssen Sie aber nicht warten – Sie können Ihr Produkt bereits in Verkehr bringen. In diesem Artikel erfahren Sie, wie Sie die Risikoklasse Ihrer Medizinprodukt-Software bestimmen: Klassifizierung von Software-Medizinprodukten: MDR Leitfaden
2.2 Ihre EU-Konformitätserklärung
Vor dem Markteintritt erklären Sie als Hersteller die Konformität Ihres Produkts schriftlich. Dieses Dokument beinhaltet alle Informationen, die in Anhang IV der MDR definiert sind. Eine entsprechende Vorlage für die EU-Konformitätserklärung können Sie hier herunterladen.
EU-Konformitätserklärung – kostenloser Download
Wir senden Ihnen eine Vorlage für die EU-Konformitätserklärung kostenlos per E-Mail zu. Tragen Sie dazu einfach Ihre E-Mail-Adresse ein.
2.3 Registrierung des Medizinprodukts: EUDAMED und DMIDS
Muss ich mein Produkt beim BfArM (DMIDS) oder in der EUDAMED-Datenbank anmelden? Oder sogar in beiden Systemen? Diese Frage stellt sich im Moment allen Medizinprodukt-Herstellern, da geplant ist, dass EUDAMED das DMIDS in Zukunft ablöst. Im Moment ist sie recht schnell beantwortet: DMIDS ist Pflicht, EUDAMED freiwillig. Hinweis: Dieser Artikel beleuchtet den Stand vom 25.03.2022. Zwischenzeitliche Änderungen konnten nicht berücksichtigt werden.
3. Rahmenbedingungen für die Zulassung von Software-Medizinprodukten
3.1 Wie lange dauert die Zulassung?
Der Zeitaufwand für die Zertifizierung Ihres Medizinprodukts kann natürlich nicht pauschal bestimmt werden. Dennoch gibt es ein paar wichtige Parameter, die Sie in der Planung berücksichtigen müssen. Der in Klammern angegebene Zeitrahmen ist als Faustregel zu betrachten – in der Realität können diese Angaben auch schwanken:
- Aufbau eines Qualitätsmanagementsystems (Einige Monate)
- Produktentwicklung & Erstellung der technischen Dokumentation (Einige Monate bis Jahre)
- Klinische Prüfung, falls notwendig (Einige Monate bis Jahre)
- Für Klasse IIa oder höher:
- Suche nach einer benannten Stelle (bis zu 6 Monate)
- Prüfung der technischen Dokumentation und des QMS durch die benannte Stelle (ca. 3-12 Monate)Das heißt aber nicht, dass Sie für ein Klasse I Produkt nichts machen müssen – Sie müssen trotzdem ein QMS aufbauen und die technische Dokumentation erstellen. Dies wird aber nicht von einer benannten Stelle überprüft.
Bestimmt haben Sie gemerkt, dass die Notwendigkeit einer benannten Stelle der wichtigste Zeitfaktor ist. Das bedeutet, dass die Zulassung von Klasse I Medizinprodukten vergleichsweise kaum Zeit in Anspruch nimmt. Tipp: Wenn Sie eine benannte Stelle benötigen, können folgende Maßnahmen dazu beitragen, den Zeitaufwand möglichst gering zu halten:
- Kümmern Sie sich möglichst früh um einen Termin bei einer benannten Stelle zur Prüfung der technischen Dokumentation für Ihr Produkt. Natürlich kann nicht auf den Tag genau abgeschätzt werden, wann Ihre technische Dokumentation final ist. Aber es ist besser, einen geplanten Termin zu vereinbaren und diesen notfalls zu verschieben, als gar keinen zu haben. Vergessen Sie aber nicht, Ihre benannte Stelle unverzüglich darüber zu informieren, falls Sie den Termin nicht einhalten können.
- Entscheiden Sie sich für eine benannte Stelle, die eine gewisse Affinität für Software-Medizinprodukte aufweist. Das reduziert Kommunikationsschwierigkeiten und beugt unpraktischen Anpassungen Ihres QMS und anderer Dokumente vor.
- Achten Sie darauf, eine möglichst fehlerfreie technische Dokumentation einzureichen. Insbesondere Flüchtigkeitsfehler und offensichtliche Unstimmigkeiten sollten Sie vorab selbst beseitigen. Denn dadurch verkleinert sich die Zahl der Feedbackschleifen, welche in der Regel jeweils mehrere Tage oder Wochen dauern.
Insgesamt sollten Sie für die Prüfung der technischen Dokumentation und die Auditierung des Qualitätsmanagementsystems mehrere Monate einplanen.
3.2 Wie viel kostet die Zulassung?
Die Kosten für die Medizinprodukt-Zertifizierung können stark variieren, daher listen wir hier die Kostenpunkte auf, die üblicherweise am stärksten ins Gewicht fallen. Grundsätzlich lässt sich auch hier sagen: Mit einem Klasse I Produkt kommen sie am günstigsten weg. Denn ab Klasse IIa benötigen Sie eine benannte Stelle für die Konformitätsbewertung Ihrer Software, welche die Kosten natürlich erhöht. Nennenswert im Allgemeinen sind an dieser Stelle die Kosten für
- Beratungsleistungen beim Aufbau eines Qualitätsmanagementsystems
- UDI-Nummern für Produkte (üblicherweise wenige 100€/Jahr)
- Klinische Prüfung, falls notwendig (mehrere 10.000€ oder mehr)
- (nur für Klasse IIa oder höher:) die benannte Stelle (ca. 30.000 – 35.000€). Hier kommt es stark darauf an, wie “zufrieden” die benannte Stelle mit Ihrer technischen Dokumentation und dem Qualitätsmanagementsystem ist. Je mehr Aufwand (z.B. Feedbackschleifen) vonseiten der benannten Stelle notwendig ist, desto höher werden die Kosten. Auch die Unternehmensgröße und die Anzahl der Produkte sind ein entscheidender Faktor für die Kosten.
Wenn wir schon von Kosten sprechen, müssen wir neben den finanziellen Aufwänden natürlich auch den Faktor Zeit betrachten. Zum einen dauert der Aufbau eines Qualitätsmanagementsystems auf jeden Fall zumindest einige Monate. Dadurch entgehen Ihnen vermutlich auf kurze Sicht Umsätze – auch wenn Sie ein konformes Qualitätsmanagementsystem natürlich als sinnvolles Investment in die Zukunft sehen müssen. Schließlich ist ein solches das Fundament eines jeden Software-Medizinproduktherstellers. Ein weiterer Zeitfresser betrifft Hersteller von Produkten der Klasse IIa oder höher: eine benannte Stelle blockiert den Markteintritt von Medizinprdoukten immer um einige Monate. Das heißt, dass Sie nichts an dem Produkt verdienen können, bevor die Zertifizierung durch die benannte Stelle abgeschlossen ist. Überlegen Sie sich also vorab, ob diese Kosten für Ihr Unternehmen realistisch zu tragen sind. Insbesondere Startups unterschätzen den Aufwand oft und haben häufig nicht die notwendigen Ressourcen, um diese “umsatzfreie” Zeit zu überstehen.
4. Fazit
Als wichtigste Kernaussage lässt sich festhalten: Aufwand, Dauer und Kosten für die Zulassung Ihrer Medizinprodukt-Software stehen und fallen mit der Risikoklasse. Für alle Klassen gilt es aber, ein Qualitätsmangamentsystem aufzubauen und die technische Dokumentation für das Produkt anzufertigen. Der große Unterschied liegt darin, dass Sie als Hersteller ab der Risikoklasse IIa weitaus intensiver unter die Lupe genommen werden. Hersteller von Medizinprodukten der Klasse I werden mit ganz viel Glück erst viele Monate nach Markteintritt von ihrer Aufsichtsbehörde überprüft (und dann oft nur oberflächlich), wohingegen andere Hersteller sich von einer benannten Stelle zertifizieren lassen müssen. Diese überprüft sowohl die technische Dokumentation Ihres Produkts, als auch Ihr Qualitätsmanagementsystem. Bei den Kosten für die benannte Stelle sollten Sie mit etwa 30.000-35.000€ rechnen (je nach Aufwand können diese Kosten natürlich weitaus höher liegen) – vergessen Sie aber nicht die großen internen Aufwände, die für den Aufbau eines Qualitätsmanagementsystems betrieben werden müssen! Hier müssen Sie bestimmt einige Monate und viel Arbeitszeit investieren. Unterschätzen Sie diese Kosten nicht und seien Sie sich bewusst, dass es im Zuge der Medizinproduktzulassung auch zu unvorhergesehenen Verzögerungen von mehreren Monaten kommen kann, bevor Sie mit Ihrem Produkt erste Umsätze generieren. Folgende Artikel sind für Sie ebenfalls relevant, wenn Sie die Entwicklung eines Software-Medizinprodukts planen:
- Formulierung der Zweckbestimmung für (Software-)Medizinprodukte
- Leitfaden: Ist Ihre App ein Medizinprodukt?
- Klassifizierung von Software-Medizinprodukten: MDR-Leitfaden
Noch mehr Artikel finden Sie in unserem Blog.
The post Zulassung & Zertifizierung von Software-Medizinprodukten (MDR) appeared first on QuickBird Medical.
]]>The post IEC 62304: Software-Lebenszyklus-Prozesse von Medizinprodukten appeared first on QuickBird Medical.
]]>An der Norm IEC 62304 kommen Sie schwer vorbei, falls Sie eine Medical App bzw. Medical Software für den europäischen Markt entwickeln möchten. Und das ist auch gut so: die IEC 62304 ist ein nützliches Werkzeug, um die Konformität Ihres Software-gestützten Medizinprodukts mit der MDR (Medical Device Regulation) oder MDD (Medical Device Directive) nachzuweisen.
Falls Sie die Entwicklung einer Medical App oder Medical Software planen, finden Sie hier einen Leitfaden, der Sie durch alle Inhalte der IEC 62304 Schritt für Schritt hindurchführt. Im Internet gibt es viele Artikel zur IEC 62304, jedoch greift sich meist jeder Artikel nur einen Teilaspekt heraus. Als Hersteller ist es daher schwer einen Überblick zu bekommen, was denn nun alles zu beachten ist. Dieser Artikel soll ihnen dieses Gesamtbild einmal aufmalen. Wir gehen jeden Abschnitt der IEC 62304 durch, und fassen ihn in verständlicher Weise mit veranschaulichenden Beispielen zusammen.
Inhaltsverzeichnis
- 1. Überblick über die IEC 62304
- 2. Anwendungsbereich (1.), Normative Verweisungen (2.) und Begriffe (3.)
- 3. Allgemeine Anforderungen (4.)
- 4. Software-Entwicklungs-Prozess (5.)
- 4.1 Planung der Software-Entwicklung (5.1)
- 4.2 Analyse der Software-Anforderungen (5.2)
- 4.3 Design der Software-Architektur (5.3)
- 4.4 Detailliertes Software-Design (5.4)
- 4.5 Implementierung der Software-Einheiten (5.5)
- 4.6 Software-Integration und -Integrationsprüfung (5.6)
- 4.7 Prüfung des Software-Systems (5.7)
- 4.8 Software-Freigabe (5.8)
- 5. Weitere Prozesse der IEC 62304
- 6. Weitere wichtige Normen
- 7. Fazit
1. Überblick über die IEC 62304
Eine Software/App, die eine medizinische Zweckbestimmung hat, ist nach MDR/MDD ein reguliertes Medizinprodukt (lesen Sie hier, ob das speziell für Ihre Software gilt). Im Rahmen des Digitale-Versorgung-Gesetz (DVG) muss ihre digitale Gesundheitsanwendung (DiGA) sogar ein Medizinprodukt sein.
Die IEC 62304 kann für regulierte Medizinprodukt-Software verwendet werden, um die Konformität mit Anforderungen z.B. der MDR oder MDD nachzuweisen. Die Anwendung der Norm im Rahmen der MDR/MDD ist theoretisch freiwillig. Sie sind dazu nicht gesetzlich verpflichtet. Die Anforderungen der MDR/MDD ohne eine derartige Norm nachzuweisen ist jedoch schwierig und bedarf am Ende vermutlich mehr Aufwand und Überzeugungsarbeit bei den Auditoren. Falls Sie eine Software/App als Medizinprodukt herstellen, empfiehlt es sich daher von der IEC 62304 Gebrauch zu machen.
Ziel der IEC 62304 ist, dass die entwickelte Medical App bzw. Medical Software innerhalb eines durchdachten Prozesses umgesetzt wird. Dabei sollen Dinge vermieden werden, die man z.B. häufig in anderen Software-Projekten findet:
- Änderungen an der Software werden umgesetzt ohne diese ausreichend zu testen
- Implikationen von bestimmten Software-Funktionen werden nicht auf Risiken untersucht. Die Implikationen werden erst bei Verwendung durch echte Nutzer klar.
- Es existiert keine durchdachte Software-Architektur. Spätere Änderungen führen daher meist zu unerwarteten Problemen.
- Aufgrund von mangelnder Dokumentation und unstrukturierter Arbeitsweise verstehen Entwickler den Code selbst nicht mehr. Sie machen daher mehr Fehler bei der Entwicklung.
- Die Software-Stände werden nicht innerhalb eines Versionierung-Systems gepflegt. Es ist daher nachträglich nur sehr aufwändig möglich festzustellen, welche Änderung einen bestimmten Fehler verursacht hat. Das Zurücksetzen auf frühere Zustände der Software ist nicht möglich.
- Anforderungen der Software werden nicht geplant. Die Entwickler implementieren die falschen Funktionen, und setzen diese nicht im Sinne des Nutzers um. Das Produkt erfüllt am Ende nicht den geplanten Zweck.
Dies sind nur einige Beispiel-Szenarien, welche die IEC 62304 zu verhindern versucht.
Wichtig zu erwähnen ist auch, dass Sie die Norm (wie auch andere ISO/IEC-Normen) kaufen müssen. Sie können die Normen z.B. bei Beuth, iso.org oder, deutlich günstiger, bei evs.ee (ca. 10% des Preises im Vergleich zu Beuth/iso.org) erwerben.
Um Ihnen vorab einen Überblick über alle Inhalte der IEC 62304 zu geben, sehen Sie hier das Inhaltsverzeichnis der Norm auf der obersten Ebene:
- Anwendungsbereich
- Normative Verweisungen
- Begriffe
- Allgemeine Anforderungen
- Software-Entwicklungs-Prozess
- Software-Wartungs-Prozess
- Software-Risikomanagement-Prozess
- Software-Konfigurationsmanagement-Prozess
- Problemlösungs-Prozess für Software
In den folgenden Kapiteln fassen wir jeden dieser Abschnitte und die zugehörigen Anforderungen prägnant für Sie zusammen.
2. Anwendungsbereich (1.), Normative Verweisungen (2.) und Begriffe (3.)
Dem eigentlichen Hauptteil aller Anforderungen der IEC 62304 gehen drei kurze Kapitel voran:
Im ersten Kapitel wird das Anwendungsgebiet der IEC 62304 spezifiziert. Zusammengefasst gilt die Norm für die Entwicklung und Wartung von Medizinprodukt-Software. Die Software kann entweder ein eigenständiges Medizinprodukt sein, oder ein eingebetteter Bestandteil eines anderen Medizinprodukts (z.B. embedded Software).
Im zweiten Kapitel finden Sie normative Verweisungen. Unter normativ versteht man hier, dass die referenzierten Dokumente für die Anwendung der IEC 62304 erforderlich sind. Hier wird nur die ISO 14971 (Risikomanagement von Medizinprodukten) genannt. Dazu später mehr.
Im dritten Kapitel werden die wichtigsten genutzten Begriffe innerhalb der IEC 62304 definiert. Diese sind für die korrekte Interpretation der Norm wichtig. Was z.B. eine „Aktivität“ von einem „Prozess“ unterscheidet, mag nicht jedem Leser klar sein (Prozess = Reihe von verbundenen Aktivitäten, die Eingaben in Ergebnisse umwandeln).
3. Allgemeine Anforderungen (4.)
Bevor wir uns den Kern-Aspekt der IEC 62304 ansehen, den Software-Entwicklungs-Prozess, sollten wir einen Blick auf die sog. allgemeinen Anforderungen werfen.
Der Hersteller muss …
- … ein Qualitätsmanagement-System führen. Dies kann z.B. durch die Anwendung der ISO 13485 (Norm für Qualitätsmanagement-Systeme von Medizinprodukten) nachgewiesen werden. (4.1)
- … einen Risikomanagement-Prozess nach ISO 14971 anwenden. (4.2)
- … jedem Software-System eine Software-Sicherheitsklasse (A, B oder C) zuweisen (4.3)
Bezüglich des Qualitätsmanagement-Systems und dem geforderten Risikomanagement sollten Sie einen Blick in die jeweiligen referenzierten Normen (ISO 13485 und ISO 14971) werfen. Diese Normen behandeln wir nicht innerhalb dieses Artikels.
Auf den sehr einflussreichen Punkt der Software-Sicherheitsklassifizierung gehen wir hier ein:
3.1 Software-Sicherheits-Klassifizierung (4.3)
Sie müssen jedem Software-System eine Sicherheitsklasse (A, B oder C) zuordnen. Je nach Sicherheitsklasse fordert die IEC 62304 mehr (Sicherheitsklasse B oder C) oder weniger (Sicherheitsklasse A) von Ihnen und Ihren Prozessen.
Es steht Ihnen hierbei in gewisser Weise frei, in wie viele Systeme Sie Ihre Software unterteilen möchten. Der Einfachheit halber bestehen viele Medical Apps aus nur genau einem Software-System. Falls Sie aber z.B. einen nicht sicherheitskritischen Teil sowie einen sehr sicherheitskritischen Teil in Ihrer Anwendung haben, könnten Sie hierfür jeweils ein Software-System der Klasse A und ein Software-System der Klasse C aufbauen. Diese Systeme müssen natürlich entsprechend technisch getrennt und unabhängig voneinander sein.
Die Klassifizierungsregeln sind in der folgenden Abbildung prägnant zusammengefasst. Im Zweifel sollten Sie einen zusätzlichen Blick in das entsprechende Kapitel (4.3) der IEC 62304 werfen.

Entscheidungsbaum zur Software-Sicherheitsklasse nach IEC 62304: Zum Vergrößern klicken
4. Software-Entwicklungs-Prozess (5.)
Der zentrale Inhalt der IEC 62304 ist die Forderung nach einem definierten Software-Entwicklungs-Prozess. Dieser Prozess besteht aus einer Anzahl von Aktivitäten, die wir hier näher erläutern.
Die geforderten Aktivitäten sind in der folgenden Abbildung dargestellt:

Software-Entwicklungs-Prozess nach IEC 62304: Zum Vergrößern klicken
Der Software-Entwicklungs-Prozess besteht also aus den folgenden Kernaktivitäten:
- Planung der Software-Entwicklung (5.1)
- Analyse der Software-Anforderungen (5.2)
- Design der Software-Architektur (5.3)
- Detailliertes Software-Design (5.4)
- Implementierung der Software-Einheiten (5.5)
- Software-Integration und -Integrationsprüfung (5.6)
- Prüfung des Software- Systems (5.7)
- Software-Freigabe (5.8)
Es fällt schnell auf, dass sich dieser Prozess stark am V-Modell der Software-Entwicklung orientiert. Es ist jedoch wichtig zu beachten, dass die IEC 62304 nicht explizit ein bestimmtes Vorgehensmodell während der Software-Entwicklung fordert. Dem Hersteller steht frei, ob er ein V-Modell, agile Entwicklung oder das Wasserfall-Modell anwendet.
Im V-Modell würden Sie die genannten Aktivitäten über den gesamten Entwicklungszeitraum der Medical App verteilen. Bei agiler Entwicklung würde z.B. jeweils jeder Sprint die genannten Aktivitäten beinhalten (zumindest einen Teil davon). Jeder Sprint wäre dann ein eigenes kleines V.
In den folgenden Abschnitten geben wir einen Überblick über jede der genannten Aktivitäten:
4.1 Planung der Software-Entwicklung (5.1)
Sie müssen einen Software-Entwicklungsplan erstellen. Dieser Plan beschreibt im Grunde wie genau bei der Software-Entwicklung zu arbeiten ist. Der Software-Entwicklungsplan beschreibt folgende Punkte (oder referenziert diese in anderen Dokumenten):
- Prozesse der Software-Entwicklung
- zu liefernde Ergebnisse und Dokumente
- Beschreibung der Rückverfolgbarkeit zwischen Anforderungen, Prüfungen und Risikokontrollmaßnahmen (Stichwort: Traceability)
- Konfigurations- und Änderungsmanagement
- Problembehandlung- und Lösung
- Software-Integration und Integrationsprüfung
- Software-Verifizierung
- Software-Risikomanagement
- Dokumentation
Er ist also eine Art Handbuch für den gesamten Prozess der Software-Entwicklung.
4.2 Analyse der Software-Anforderungen (5.2)
Sie müssen alle Anforderungen an Ihre Software dokumentieren. Dazu gehören funktionale Anforderungen sowie nicht-funktionale Anforderungen. Genauer gesagt müssen Sie u.a. Folgendes dokumentieren:
- Anforderungen an Funktionalität und Leistungsfähigkeit
- Ein- und Ausgaben des Software-Systems
- Schnittstellen zwischen dem Software-System und anderen Systemen
- Software-gesteuerte Alarme, Warnungen und Anwender-Meldungen
- Anforderungen für die Datensicherheit
- Anforderungen für die Benutzerschnittstelle
- Datendefinition und Datenbank-Anforderungen
- Anforderungen für Installation und Abnahme
- Anforderungen für Betrieb und Wartung
- Anforderungen bezüglich IT-Netzwerkaspekten (z.B. Handhabung, wenn Netzwerk nicht verfügbar ist, Netzwerk-Protokolle, etc.)
- regulatorische Anforderungen
- Einbeziehung von Risikobeherrschungsmaßnahmen (zur Verminderung oder Eliminierung von identifizierten Risiken)
Diese Punkte können sich auch gegenseitig überlappen. Alle dokumentierten Software-Anforderungen müssen (nach der Implementierung) verifiziert werden. Die Verifizierung lässt sich zusammenfassen als Prüfung, ob alle Anforderungen erfüllt sind. Dafür müssen alle Anforderungen so formuliert werden, dass eine Prüfung anhand von definierten Prüfkriterien möglich ist.
4.3 Design der Software-Architektur (5.3)
Innerhalb von Software-Systemen der Sicherheitsklasse B oder C müssen Sie vor der Implementierung die geplante Software-Architektur dokumentieren. Diese inkludiert auch Schnittstellen zwischen einzelnen Software-Komponenten und z.B. externen Systemen. Sie können dies z.B. grafisch mit Hilfe von UML oder C4 umsetzen. Dabei können Sie z.B. die Software-Architektur selbst mit Hilfe von UML-Klassendiagrammen beschreiben, und einzelne Kern-Abläufe z.B. mit Hilfe eines Sequenzdiagramms.
Eine Medical App bzw. Medical Software kommt selten ohne Abhängigkeiten zu externen Software-Paketen (z.B. für die Anzeige oder Speicherung von Daten) aus. Diese Software-Pakete (bzw. Software-Komponenten) bezeichnet man als SOUP (Software Of Unknow Provenance). Derartige Komponenten wurden normalerweise nicht vom eigenen Team entwickelt und wurden nicht innerhalb eines geeigneten Qualitätsmanagement-Systems dokumentiert. Sie müssen daher u.a. selbst Funktions- und Leistungsanforderungen für die SOUP-Komponente festlegen. Konkret schreiben Sie dafür eine Liste aller Abhängigkeiten (Dependencies), und legen für jede Abhängigkeit fest, ob und wie Sie diese überprüft haben. Wie detailliert Sie dies tuen müssen hängt von der Sicherheitsklasse des Systems ab.
Zuletzt müssen Sie auch die Software-Architektur verifizieren, und z.B. nachweisen, dass diese Ihre Software-Anforderungen implementiert.
4.4 Detailliertes Software-Design (5.4)
Bei Software-Design handelt es sich nicht um Design im Sinne der Nutzeroberfläche (Nutzer-Input/Output), sondern um eine Dokumentation der Software-Einheiten auf technischer Ebene (ähnlich zur Software-Architektur). Im Gegensatz zur Software-Architektur, welche die Software-Komponenten abbildet, zeigt das detaillierte Software-Design jedoch alle Software-Einheiten und ihre Beziehungen zueinander auf. Software-Design existiert also auf einem niedrigeren Abstraktionsniveau und ist somit detaillierter.
Im Gegensatz zu einer Software-Komponente definiert sich eine Software-Einheit als „nicht weiter unterteilbare“ Einheit. Eine Software-Komponente kann aus mehreren Software-Einheiten bestehen. Ein Software-System besteht aus Software-Komponenten. Als Faust- und Merk-Regel könnte man es so definieren:
Software-System ≥ Software-Komponente ≥ Software-Einheit
Dieses Design muss detailliert genug sein, um die korrekte Implementierung zu ermöglichen. Außerdem muss es natürlich in Harmonie mit der Software-Architektur stehen, frei von Widersprüchen.
4.5 Implementierung der Software-Einheiten (5.5)
Nach ausführlicher Dokumentation der genannten Punkte, kommt nun endlich der Teil, auf den wohl jeder Software-Entwickler gewartet hat: die eigentliche Implementierung der Software-Einheiten.
Für Software-Systeme der Sicherheitsklassen B oder C müssen außerdem alle Software-Einheiten einzeln verifiziert werden und Akzeptanzkriterien festgelegt werden. Für die Verifizierung können Sie z.B. auch auf Ihre Codierungs-Normen verweisen (Coding Guidelines). Diese definieren z.B. wie viele Zeilen Code Sie maximal pro Software-Methode und Software-Klasse zulassen (um Übersicht zu gewährleisten), die zu erreichende Unit-Test-Coverage und sonstige Programmiersprachen-abhängige Vorschriften. Um die Einhaltung Ihrer Codierungs-Normen zu überprüfen bietet sich ein Tool für statische Code-Analyse an (in Kombination mit einem Linter, der Verstöße sofort für den Entwickler im Code markiert).
Außerdem sind Code-Reviews ideal, um die Einhaltung der Anforderungen, Akzeptanz-Kriterien und Software-Metriken zu überprüfen. Ein Entwickler prüft den Code des anderen Entwicklers vor Freigabe (bzw. vor jedem „Git-merge“) und hinterlässt Kommentare mit Nachbesserungs-Vorschlägen. Diese Kommentare sind nachträglich ein idealer Nachweis, um schriftlich zu belegen, dass sie die Prozesse der IEC 62304 einhalten. Dies geschieht normalerweise mit Hilfe von Code-Review-Funktionalitäten auf z.B. GitHub, GitLab oder Bitbucket.
4.6 Software-Integration und -Integrationsprüfung (5.6)
Nach der Implementierung der einzelnen Software-Einheiten müssen diese natürlich im selben System landen. Für Software-Systeme der Sicherheitsklassen B oder C müssen Sie diese nach einem dokumentierten Integrationsplan in das Gesamtsystem integrieren. Anschließend muss geprüft werden, ob die Integration erfolgreich war, oder ob z.B. Probleme entstanden sind.
Konkret kann dies entweder durch manuelle Tests z.B. eines QA-Experten (der Titel ist nicht entscheidend) geschehen oder durch automatisierte Integration-Tests in der Software selbst.
4.7 Prüfung des Software-Systems (5.7)
Sie müssen eine Reihe von Prüfungen festlegen und durchführen, die alle Software-Anforderungen abdecken. Dafür geben Sie u.a. Eingabe-Werte und erwartete Ausgabewerte an, setzen Pass/Fail-Kriterien und beschreiben generell Ihre Verfahren zur Software-System-Prüfung.
Diese Prüfungen können natürlich sowohl (menschliche) manuelle Tests als auch automatisierte Tests (UI-Tests, Integration-Tests, Unit-Tests, etc.) jeglicher Art beinhalten.
Gleichzeitig müssen diese Tests so dokumentiert werden, dass Sie wiederholbar sind. Dafür müssen Sie bei der Prüfung z.B. Ihre Hardware- und Software-Konfiguration, Prüfwerkzeuge und die Version der geprüften Software dokumentieren. Außerdem empfiehlt sich eine Dokumentation der Test-Schritte, um Wiederholbarkeit zu gewährleisten.
Das kann z.B. so aussehen:
- Öffne die App – Erwartetes Ergebnis: Startbildschirm mit „Messen“-Knopf ist sichtbar
- Klicke auf den „Messen“-Knopf – Erwartetes Ergebnis: die Kamera öffnet sich. Der „Aufnahme“ Button ist sichtbar.
- Klicke auf den „Aufnahme“-Knopf – Erwartetes Ergebnis: Ein Ladebalken öffnet sich.
- …
Derartige Testabläufe können Sie entweder in Word-Dokumenten dokumentieren, oder Tools wie Jira (jedes Ticket ist ein Test-Fall) oder TestRail benutzen.
4.8 Software-Freigabe (5.8)
Nach erfolgreicher Verifizierung der Software kann die Software freigegeben werden. Sie wird in ein ausführbares Format kompiliert. Restliche Anomalien werden dokumentiert. Die fertige Software wird mitsamt der zugehörigen Dokumentation archiviert und kann ausgeliefert werden. Pragmatisch betrachtet reicht hier z.B. ein Ordner für jede neue freigegebene Version: Quellcode, kompilierter, ausführbarer Code (z.B. als APK-Datei für Android-Systeme) und alle Build-Tools, die zur erneuten Erstellung dieser ausführbaren Software-Version nötig sind (z.B. die Installationsdatei der konkreten Version der Entwicklungsumgebung).
Bei der Auslieferung an Ihre Nutzer muss sichergestellt werden, dass auf dem Weg keine unerwünschten Änderungen/Verfälschungen der Software mehr passieren können. Bei Software impliziert dies vor allem Sicherheitsvorkehrungen, damit keine Hacker ihren fertigen Software-Release manipulieren können (und so z.B. Schadcode einschleusen).
Es kann natürlich sein, dass Ihre Software und Ihr Qualitätsmanagement-System z.B. im Rahmen der MDR vor der Freigabe an Nutzer erst von einer benannten Stelle geprüft werden muss. Das hängt von der Gesetzgebung Ihrer Zielmärkte ab. Die IEC 62304 ist unabhängig davon und schlägt nur einen Prozess vor, den Sie zum Nachweis der Konformität umsetzen können.
Falls Ihr Produkt z.B. schon auf dem europäischen Markt ist, müssen Sie für jede Änderung entscheiden, ob diese „signifikant“ ist. Wenn es sich um eine „signifikante Änderung“ handelt, muss Ihre benannte Stelle die Änderung vor der Freigabe absegnen. Diese Forderung ist jedoch Teil der MDR bzw. MDD, und nicht Teil der IEC 62304.
5. Weitere Prozesse der IEC 62304
Der Software-Entwicklungs-Prozess ist mit Abstand der umfangreichste Teil der IEC 62304. Darüber hinaus müssen Sie allerdings noch vier weitere Prozesse aufbauen: für Software-Wartung, Software-Risikomanagement, Software-Konfigurationsmanagement und Problemlösung. Auf jeden dieser Prozesse gehen wir in Folgendem kurz ein:
5.1 Software-Wartungs-Prozess (6.)
Für den Betrieb der Software muss ein Software-Wartungs-Prozess definiert werden. Dafür wird zuerst ein Software-Wartungs-Plan aufgesetzt. Dieser beschreibt wie nach Freigabe der Medical App bzw. Medical Software mit Rückmeldungen von Nutzern oder anderen Parteien umgegangen werden soll. Es werden z.B. Kriterien dafür definiert, ab wann eine Rückmeldung als Problem zu betrachten ist. Im Falle eines Problems kommt der Problemlösungs-Prozess zum Einsatz.
Außerdem muss jede Änderungsanforderung an der Software analysiert und genehmigt werden. Nach Genehmigung der Änderung werden diese mit Hilfe des Software-Entwicklungs-Prozesses wie gewohnt implementiert.
Während der Software-Wartung kommt weiterhin ein definierter Risikomanagment-Prozess zum Einsatz (siehe unten). Jede Änderung durchläuft das Risikomanagement und wird auf mögliche Gefährdungen untersucht.
Einen Überblick der Software-Wartungs-Prozesse und Aktivitäten sehen Sie hier:

Software-Wartungs-Prozess nach IEC 62304: Zum Vergrößern klicken
Wie man sieht ist der Software-Wartungs-Prozess lediglich eine leichte Abwandlung des Software-Entwicklungs-Prozesses.
5.2 Software-Risikomanagement-Prozess (7.)
Die einzige weitere Norm, welche von der IEC 62304 vorrausgesetzt wird, ist die ISO 14971. Diese beschreibt das Risikomanagement von Medizinprodukten. Zusammengefasst müssen Sie Ihre Medical Software daher auf mögliche Gefährdungen (= Schadensquellen) und Gefährdungssituationen untersuchen. Das mit diesen verbundene Risiko muss eingeschätzt werden. Auf dieser Basis wird nun evaluiert, ob ein Risiko akzeptabel ist oder es z.B. durch Risikokontrollmaßnahmen verringert werden muss. Die ISO 14971 gilt auch für Hardware-Medizinprodukte und nimmt daher nicht explizit Stellung zu Problematiken im Rahmen der Software. Die IEC 62304 verweist für diesen Prozess hauptsächlich auf die ISO 13485 und stellt keine nennenswerten Zusatzanforderungen.
Hier sehen Sie einen Überblick über alle Prozesse und Aktivitäten im Rahmen des Risikomanagements nach ISO 14971:

Software-Risikomanagement-Prozess nach ISO 14971: Zum Vergrößern klicken
Im Rahmen des Risikomanagements werden Sie dann z.B. eine Failure Mode and Effects Analysis-Tabelle (FMEA) erstellen oder ein Fault-Tree-Analyse (FTA) durchführen.
5.3 Software-Konfigurationsmanagement-Prozess (8.)
Dieser Prozess definiert, wie Änderungen und Freigaben in der Software kontrolliert bzw. protokolliert werden.
Dies kann zum Beispiel durch ein Software-Versionierung-Tool wie Git geschehen. Änderungen werden als einzelne „Commits“ (Änderungspakete) festgehalten, und freigegebene Versionen werden als solche markiert (z.B. durch einen „Release“-Tag). Außerdem fordert dieser Abschnitt, dass die System-Konfiguration und mögliche SOUPs systematisch dokumentiert werden.
Der zweite Teil des Prozesses beschäftigt sich mit der sog. Änderungskontrolle. Jegliche Änderungen dürfen nur nach vorheriger Genehmigung einer entsprechenden Änderungsanforderung implementiert werden. In diesem Rahmen müssen Sie bei Änderungen alle Aktivitäten identifizieren, die wiederholt werden müssen. Dies können Schritte des Software-Entwicklungs-Prozesses sein, oder auch die Software-Sicherheitsklassifizierung der Software-Systeme.
5.4 Problemlösungsprozess für Software (9.)
Für jedes neue Problem, das Sie in Ihrer Medical Software identifizieren, müssen Sie einen Problembericht anlegen. Dieser Bericht enthält dann z.B. eine Einschätzung, wie kritisch das Problem ist, sowie jegliche Informationen, die bei der Problemlösung helfen könnten (z.B. betroffene Geräte oder Anweisungen für die Reproduktion des Problems).
Außerdem müssen Sie für jedes Problem wenn möglich Ursachen identifizieren. Das Risikomanagement wird für das Problem neu angestoßen. Und schlussendlich wird eine Änderungsanforderung für die Lösung des Problems gestellt. Nach der Implementierung dieser Änderungsanforderung muss diese natürlich wieder verifiziert werden:
Wurde das Problem gelöst oder besteht es weiterhin? Wurden dadurch zusätzliche Probleme verursacht?
Derartige Fragen müssen Sie sich nach jeder Änderungsanforderung stellen.
6. Weitere wichtige Normen
Dies war ein Überblick über alle Abschnitte der IEC 62304. Falls Sie eine medizinische Software bzw. App auf den europäischen Markt bringen möchten, sollten Sie sich auch noch folgende Normen ansehen:
- IEC 62366 (Gebrauchstauglichkeit)
- ISO 13485 (Qualitätsmanagement)
- ISO 14971 (Risikomanagement)
- IEC 82304 (Gesundheitssoftware Produktsicherheit – sinnvolle Erweiterung der IEC 62304)
Mehr zu diesen Normen finden Sie auch in unserem Leitfaden für die Entwicklung von Medical Apps.
7. Fazit
Die IEC 62304 fordert grundsätzlich viele sinnvolle Dinge, die ein gutes Software-Team sowieso schon befolgen sollte. Trotzdem wird sich ein Team darauf einstellen müssen deutlich mehr Dokumentation und Aufzeichnungen als bei herkömmlicher „Nicht-Medizinprodukt“-Software zur erstellen. Viele Software-Entwickler sind es außerdem nicht gewöhnt nach sehr strikt definierten Prozessen zu arbeiten. Das Entwickeln von medizinischer Software nach IEC 62304 bringt also durchaus weitreichende Änderungen mit sich. Falls Sie zusätzlich die Einbindung von künstlicher Intelligenz (KI) planen, sollten Sie dies in Ihrem Software-Lebenszyklus berücksichtigen (mehr dazu in unserem KI-Leitfaden).
Wir haben versucht Ihnen im Rahmen dieses Artikels einen vollständigen Überblick über alle Punkte der IEC 62304 zu geben. Eben dieser Überblick ist nötig, wenn man sich anfängt mit der Entwicklung medizinischer Software auseinander zu setzen. Weitere Anforderungen an die Entwicklung von Software als Medizinprodukt finden Sie in unserem Leitfaden zur Entwicklung von Medical Apps. Falls Sie eine digitale Gesundheitsanwendung (DiGA) im Rahmen des DVG planen, finden Sie alle Anforderungen in unserem Leitfaden zur DiGAV.
The post IEC 62304: Software-Lebenszyklus-Prozesse von Medizinprodukten appeared first on QuickBird Medical.
]]>The post Corona Apps & Contact Tracing: Alles, was Sie wissen müssen appeared first on QuickBird Medical.
]]>Besonders viel Hoffnung wird dabei auf sogenannte „Corona-Apps“ gelegt. Sie sollen dabei helfen, das Virus trotz Lockerungsmaßnahmen unter Kontrolle zu halten und dessen Verbreitung zu verlangsamen.
Aber was genau sind diese „Corona-Apps“ oder „Contact Tracing-Apps“ eigentlich? Wie funktionieren sie und welche Unterschiede gibt es bei den Apps, die bereits in anderen Ländern eingesetzt werden? Und welche Probleme bringen diese Apps mit sich, vor allem im Datenschutz?
Unser Team entwickelt tagtäglich Medical Apps und Health Apps für Kunden im Gesundheitswesen. Auf Basis unserer Erfahrung, möchten wir hier konkrete Antworten auf diese Fragen geben.
Was ist Contact Tracing?
„Corona Apps“ basieren auf dem „Contact Tracing“. Darunter versteht man eine medizinisch-epidemiologische Methode zur Rückverfolgung von Infektionskrankheiten, wie beispielsweise COVID-19. Ziel dieser Methode ist es, mögliche Kontaktpersonen von Infizierten schnell zu identifizieren, diese darüber zu informieren und Maßnahmen einzuleiten, um die weitere Verbreitung der Krankheit zu verhindern bzw. zu verlangsamen. Es geht also vor allem darum, Infektionsketten frühzeitig zu erkennen und zu unterbrechen.

Das funktioniert natürlich auch ohne Apps. Bei der manuellen Variante der Kontaktnachverfolgung werden infizierte Personen von Mitarbeitern der Gesundheitsbehörden befragt, mit welchen anderen Personen sie in ihrer infektiösen Zeit (bis zu 21 Tage nach der Infektion) Kontakt hatten. Diese werden dann informiert, dass eine Kontaktperson positiv auf die Krankheit getestet wurde und je nach Risikostufe werden verschiedene Maßnahmen eingeleitet – etwa eine vorgeschriebene Quarantäne oder ein Test, ob sie nun selbst Träger des Virus sind.
Dieses Verfahren ist sehr aufwendig und nur dann sinnvoll, wenn die Anzahl der infizierten Personen noch relativ klein ist. Diese Schwelle wurde bei COVID-19 bereits nach kurzer Zeit überschritten und obwohl die Gesundheitsbehörden ihr Personal zurzeit stark aufstocken, wird es in naher Zukunft nicht möglich sein, die Virusverbreitung alleine mit der manuellen Kontaktnachverfolgung einzudämmen.
Wie können uns Smartphones helfen?
Durch die Verwendung moderner technischer Werkzeuge, wie Smartphones, kann ein Teil des Prozesses der Kontaktnachverfolgung automatisiert, verbessert und deutlich schneller durchgeführt werden. Vor allem einige asiatische Länder, allen voran Singapur, haben sehr schnell reagiert und Smartphone-Apps als Hilfsmittel zur Eindämmung der Corona-Pandemie eingesetzt. Andere Länder sind dem Beispiel bereits gefolgt und viele weitere planen die Einführung von Corona-Apps in den folgenden Wochen. Auch in Deutschland ist eine solche Corona-App im Gespräch.
Ziel einer solchen App ist es, genau wie bei der manuellen Kontaktnachverfolgung die unmittelbare Nähe zu einer infizierten Person festzustellen und die Nutzer vor einer möglichen Infektion zu warnen.
Dazu gehören immer drei Schritte:
1. Aufzeichnung
Kontakte zu anderen Personen müssen durchgehend aufgezeichnet werden.
2. Meldung
Eine infizierte Person muss sich sich als infiziert melden bzw. markieren.
3. Benachrichtigung
Alle Personen, die im möglichen Infektionszeitraum unmittelbaren Kontakt mit einer als infiziert gemeldeten Person hatten, werden darüber informiert.
Technologien zur Nachverfolgung von nahen Kontakten
Um einen möglichen Kontakt zu erkennen, werden verschiedenste Technologien eingesetzt: Funkzellendaten, das Global Positioning Network (GPS) und, die wohl am meisten erfolgsversprechende Technologie, Bluetooth Low Energy (BLE). Aber wie genau funktionieren diese?
Funkzellendaten
Smartphones wählen sich durchgehend über Funkmasten in das jeweilige Netz des Mobilfunkbetreibers ein. Da sich meist mehrere Mobilfunkmasten in der Umgebung befinden, können deren Positionen und die Richtung der Signale für die sogenannte Triangulation verwendet werden. Damit kann der Betreiber der Mobilfunknetze die Position eines Smartphones und folglich auch die Position dessen Nutzers auf etwa 50 Meter genau berechnen.
Diese Methode zur Positionserkennung wird bereits vielfach angewandt (z.B. zur Verfolgung von Kriminellen) und hat den Vorteil, dass dabei keine App auf den Smartphones installiert werden muss. Damit kann natürlich die Freiwilligkeit der Datenabgabe nicht gewährleistet werden und aus diesen Daten könnten sich sehr leicht komplexere Bewegungsprofile generieren lassen.
Darüber hinaus eignet sich die Genauigkeit von 50 Metern nicht wirklich, um die unmittelbare Nähe von Personen genau zu identifizieren. Daher kommt diese Technologie für die Bekämpfung der Corona-Pandemie nicht in Betracht.
Global Positioning System (GPS)
Für eine genauere Positionierung von Personen kann das sogenannte Global Positioning System (besser bekannt als GPS) verwendet werden. Damit erreicht man im Durchschnitt bereits eine Genauigkeit von nur wenigen Metern.
Allerdings ist die Genauigkeit sehr von der Umgebung abhängig, je nachdem wie stark die Satellitensignale abgeschirmt werden. Sie ist beispielsweise in einem stark bebauten Gebiet viel geringer. Außerdem funktioniert die Technologie nicht mehr, sobald sich eine Person im Inneren eines Gebäudes befindet, da dabei keine Verbindung zu den GPS-Satelliten aufgebaut werden kann.
Damit ist auch diese Technologie eher ungeeignet für die Kontaktnachverfolgung, die mit Contact Tracing-Apps ermöglicht werden soll. Trotzdem wird sie vor allem in Kombination mit anderen Technologien eingesetzt, wie beispielsweise bei den Corona-Apps in China oder Israel. Denn das GPS-Signal kann dafür verwendet werden, um infizierte Personen zu finden und diese dann zu Quarantänemaßnahmen zu zwingen. Eine solche Anwendung ist in Deutschland nicht geplant.
Bluetooth Low Energy (BLE)
Die Technologie, welche momentan als am geeignetsten für den Einsatz der Kontaktverfolgung eingestuft wird, ist Bluetooth Low Energy (BLE). Diese Funktechnik ist in den meisten modernen Smartphones verbaut und ermöglicht es diesen, mit Bluetooth-fähigen Geräten wie drahtlosen Kopfhörern oder anderen Smartphones über kurze Distanz zu kommunizieren.
BLE eignet sich somit also im Gegensatz zu den zuvor genannten Technologien nicht zur Positionsbestimmung von Nutzern, kann aber sehr gut dazu verwendet werden, andere Nutzer im nahen Umkreis zu erkennen. Mit der Signalstärke kann man zudem ungefähr feststellen, wie groß die Distanz zwischen zwei Smartphones ist.
Genau diese Funktionalität nutzen Contact Tracing-Apps aus. Die Apps laufen im Hintergrund, also auch während sich das Smartphone im Standby-Modus befindet oder andere Apps verwendet werden. Sie senden kontinuierlich Bluetooth-Signale an alle anderen Geräte in ihrer Nähe und zeichnen wiederum die empfangenen Signale der anderen Smartphones auf. Dabei wird eine Art Logbuch geführt, welche Geräte an welchem Zeitpunkt und mit welcher Signalstärke bzw. Distanz erkannt wurden.

Wird nun ein App-Nutzer positiv auf COVID-19 getestet, kann er dieses Logbuch an einen Server hochladen. Bei der Auswertung dieser Daten gibt es zwei verschiedene Ansätze:
1. Zentraler Ansatz
Der Server wertet die Daten selbstständig aus. Er ermittelt, welche Nutzer sich in der Nähe der infizierten Person aufgehalten haben und informiert diese dann über deren App.
2. Dezentraler Ansatz
Der Server schickt die Daten an alle Nutzer der App, welche diese dann lokal auf dem Smartphone auswertet.
Beide Ansätze bringen Vor- und Nachteile mit sich, die wir im Folgenden noch besprechen werden.
Der Einsatz von Bluetooth Low Energy für die Kontaktnachverfolgung gilt für Deutschland als gesichert. Auch andere europäische Länder verfolgen diese Strategie und einige arbeiten dabei zusammen an gemeinsamen Standards. Im nachfolgenden Teil dieses Artikels gehen wir deshalb davon aus, dass Bluetooth Low Energy als Technologie zur Kontaktnachverfolgung eingesetzt wird.
Auswirkungen auf den Datenschutz
Obwohl der Einsatz von Contact Tracing-Technologien langfristig Leben retten und die Lockerung von einigen Freiheitseinschränkungen ermöglichen kann, darf der Datenschutz dabei nicht komplett außer Acht gelassen werden. Es gilt bereits als bewiesen, dass durch den Einsatz von smarten und modernen Lösungen ein zu großer Eingriff in die Datenschutzrechte und die Privatsphäre der Nutzer vermieden werden kann.
Der Ablauf des zuvor beschriebenen Mechanismus kann nahezu vollständig anonym durchgeführt werden. Natürlich muss die Nutzung dieser Apps auf freiwilliger Basis stattfinden, die Tracing-Daten dürfen vor einem amtlich bestätigten positiven Testresultat ausschließlich lokal gespeichert werden und die gesendeten Bluetooth-Daten sollten anonymisiert werden. Der Chaos Computer Club hat hier bereits 10 Anforderungen an Contact Tracing-Apps definiert.
In Europa haben sich mehrere Initiativen gebildet, um Contact Tracing-Standards zu entwickeln, die zu unserer Datenschutzgrundverordnung (DSGVO) konform sind. Hierzu zählen die beiden bekanntesten Initiativen PEPP-PT (Pan-European Privacy-Preserving Proximity Tracing) und DP-3T (Decentralized Privacy-Preserving Proximity Tracing).
Beide Standards haben einige Gemeinsamkeiten. Jedes Smartphone erhält eine zufällige Identifikationsnummer und sendet diese kontinuierlich per Bluetooth an die Geräte im Umkreis. Diese ID wird nun in regelmäßigen Zeitabständen geändert. Dadurch ist es nicht möglich, einzelne Personen nach Ablauf dieses Zeitfensters wiederzuerkennen.
Das verwendete Zeitfenster wird dabei so klein gewählt, dass keine Bewegungsprofile erstellt werden können, muss aber gleichzeitig so groß sein, dass ein längeres Aufhalten in der Nähe einer Person nachgewiesen werden kann. Beispielsweise kann mit einem Zeitfenster von 15 Minuten (Empfehlung des Robert-Koch-Instituts) erkannt werden, ob ein Gerät mehrmals gesehen wurde. Dies deutet darauf hin, dass sich eine Person für einen längeren Zeitraum in der Nähe aufgehalten hat und ein erhöhtes Infektionsrisiko besteht, sofern genau diese andere Person positiv auf COVID-19 getestet wurde.
Vergleich der verschiedenen Ansätze
Die anonymen und temporären Daten der Begegnungen werden dabei in verschlüsselter Form nur lokal auf den Smartphones gespeichert. Erst, wenn es wirklich ein positives Testergebnis gab, kann ein Nutzer seine Contact-Tracing-Daten freigeben.
Damit die positiven Testergebnisse wirklich validiert werden können und es nicht zu absichtlichen Falschmeldungen kommt, müssen die positiven Tests amtlich bestätigt werden, bevor die Daten freigegeben werden können. Dazu kann die Gesundheitsbehörde beispielsweise einen Code generieren, der für die Meldung in der App benötigt wird. Dies setzt natürlich voraus, dass es dafür einen einheitlichen Standard gibt und die Gesundheitsbehörden das technische Know-How dafür besitzen.
Wie bereits erwähnt, gibt es zwei konkurrierende Varianten, deren zentraler Unterschied darin besteht, ob es einen zentralen – und somit allwissenden – Server gibt oder nicht. Beide Varianten haben Vor- und Nachteile:
Zentrale Lösung
Bei der ersten Variante (PEPP-PT ist deren prominentester Vertreter) gibt es einen zentralen Server, welcher die zufälligen Identifikationsnummern generiert und an die Smartphones weitergibt. Diese Daten geben aber noch keine Rückschlüsse auf die Identität der Kontaktpersonen.
Zu jedem Nutzer wird dabei zusätzlich ein sogenannter Push Token gespeichert. Dieser ermöglicht es dem Server, Nutzer zu benachrichtigen, die Kontakt zu einer positiv getesteten Person hatten. Man kann sich diesen Token als eine sehr schwer zu merkende, vorübergehende Telefonnummer vorstellen.
Somit sind die Identifikationsnummern natürlich nicht mehr wirklich anonym, sondern lediglich pseudonymisiert (jeder Mensch, der meine Telefonnummer kennt, kann mich womöglich auch über meine Telefonnummer identifizieren). Die Push Tokens werden von den Betriebssystemen der Smartphones generiert (meist Android oder iOS). Entsprechend findet auch die Zuweisung der Push Token an die einzelnen Nutzer auf Apples bzw. Googles Servern statt.
Wird nun ein Patient positiv getestet, kann er sein Kontaktlogbuch der vergangenen Tage auf den zentralen Server hochladen. Auf Basis dieses Logbuchs errechnet der Server die Wahrscheinlichkeit einer Infektion für jede begegnete Person und kann diese Person anhand der Push Token gezielt informieren.
Grundlage dieser Berechnung sind mehrere Parameter, unter anderem die gemessene Signalstärke zwischen beiden Smartphones, der Typ des Smartphones (verschiedene Smartphones senden unterschiedlich starke Signale aus) und das Datum des Kontakts. Der Algorithmus kann sich in dieser zentralen Variante immer wieder ändern und sich mit neu gewonnen Daten verbessern. Zusätzlich erlaubt die zentrale Lösung in einfacher Art und Weise, diese erhobenen Daten anschließend für wissenschaftliche Zwecke zu verwenden, um weitere Erkenntnisse über die Ausbreitung des Virus zu bekommen.
Der zentrale Server muss dabei nicht zwangsläufig ein einzelner Server sein. Jedes Land bzw. jede Gesundheitsbehörde kann auf eine eigene App und/oder einen eigenen Server setzen. Um dabei aber Infektionen über Ländergrenzen hinweg zu erkennen, tauschen diese Server ihre Daten untereinander über Schnittstellen aus.
Wie bereits erwähnt, ist bei dieser Variante der zentrale Server das allwissende Element. Die Logbücher werden zwar erst nach einem durchgeführten positiven Test und auf freiwilliger Basis hochgeladen, allerdings können Bewegungsprofile von Personen auch durch die Logbuch-Daten anderer Personen erstellt werden. In der Folge muss dieser zentrale Server also von einem vertrauenswürdigen Institut bereitgestellt werden muss. Der Server ist dabei natürlich ein sehr attraktives Ziel für Hacker.
Dezentrale Lösung
Die dezentrale Lösung hat als oberstes Ziel, genau dieses Problem zu lösen und ohne zentralen Server auszukommen. Ihr bekanntester Vertreter ist das DP3T-Projekt (kurz für „Decentralized Privacy-Preserving Proximity Tracing“).
Hier sollen die temporären Identifikationsnummern lokal auf dem Smartphone generiert werden. Eine Zuordnung der Nutzer auf einem zentralen Server ist damit also nicht möglich. Kommt es nun zu einem positiven Testresultat, kann der Infizierte seine Contact-Tracing-Daten für alle App-Nutzer freigeben. Die Identifikationsnummern der Kontakte werden dann an alle Nutzer der Contact-Tracing-App weitergegeben. Jedes Smartphone mit einer solchen App gleicht dann die Identikationsschlüssel der positiv getesteten Personen mit seinem lokalen Logbuch ab. Sollte dieser Vergleich eine Übereinstimmung finden – der Benutzer war also in Kontakt mit der infizierten Person – wird er von der App auf seinem Smartphone informiert.
Da der Algorithmus lokal auf dem Smartphone ausgeführt wird und die Server nur für den Datenaustausch der Smartphones verwendet werden, gibt es auch kein zentrales Angriffsziel, ein Datenmissbrauch wird stark erschwert. Dieser dezentrale Ansatz wird unter anderem vom Chaos Computer Club präferiert.
Apple und Googles Zusammenarbeit

Über 99% aller Smartphones verwenden als Betriebssystem entweder Googles Android oder Apples iOS. Die beiden Hersteller haben in einer gemeinsamen Pressemitteilung erklärt, zusammenarbeiten zu wollen, um die Kontaktnachverfolgung durch Smartphones mit einem gemeinsamen Standard zu ermöglichen.
Ziel ist es die Kompatibilität zwischen allen Smartphones, aber auch zwischen den verschiedenen Apps sicherzustellen. Das alles soll ermöglicht werden, während Datenschutz und Transparenz gewährleistet bleiben.
Beide wollen Programmierschnittstellen, sogenannte APIs, in ihren Betriebssystemen zur Verfügung stellen, um Gesundheitsbehörden (leichter) zu ermöglichen, Contact Tracing-Apps zu entwickeln. Der Zugriff zu diesen Schnittstellen wird stark eingeschränkt sein und wohl nur Regierungs-, Gesundheits- und verwandten Behörden zur Verfügung stehen.
In einem zweiten Schritt wollen Apple und Google eine umfassendere Integration in ihre Betriebssysteme einführen. Noch ist nicht genau klar, was die beiden Hersteller genau umsetzen wollen. Ziel soll es aber sein, die Funktionalität noch prominenter auf Smartphones zu präsentieren und auch Nutzern auch die Teilnahme ermöglichen, die sich keine Apps installieren wollen oder können.
Der von den Apple und Google entwickelte Standard ist nach jetzigem Stand nur mit einer dezentralen Variante vereinbar und löst dabei bereits einige komplizierte und datenschutzrelevante Themen durch den Einsatz von kryptographischen Methoden.
Auf Apples iOS-Betriebssystem ist eine Umsetzung einer Contact Tracing-App ohne Verwendung dieser neuen Schnittstellen nicht ohne Einschränkungen möglich. Die Sicherheitsmaßnahmen von iOS schränken die Bluetooth-Funktionalität von Apps im Hintergrund sehr stark ein. Somit können auch die Identifikationsnummern nicht zuverlässig ausgetauscht werden. Um diese Einschränkung zu umgehen müsste die App durchgehend geöffnet bleiben. Dies hat natürlich starke Auswirkungen auf die Akkulaufzeit des Smartphones und ist auch sonst nicht besonders praktikabel.
Die zukünftigen Anapssungen werden dieses Problem lösen. Dies zeigt jedoch auch wie abhängig alle Länder und Gesundheitsbehörden von Apple/Google bei der Umsetzung von praktikablen Lösungen sind.
Ausblick
In den folgenden Wochen werden einige weitere Länder ihre eigenen Contact Tracing-Apps auf Basis der Bluetooth-Technologie veröffentlichen. Länder wie Singapur oder Österreich haben ihre Apps bereits veröffentlicht und den Quellcode und das Protokoll frei zur Verfügung gestellt. Deutschland plant momentan die Entwicklung einer solchen App, die dem dezentralen Ansatz folgt. Andere Länder wie Frankreich arbeiten weiterhin daran, eine zentrale Lösung umsetzen und versuchen Druck auf Apple aufzubauen, um die technischen Hürden zu überwinden.
Die meisten dieser Apps sollen nach der COVID-19-Pandemie wieder zurückgenommen werden, befinden sich dann aber sicher auch weiterhin in der Schublade der Gesundheitsbehörden. Vorstellbar wäre der Einsatz solcher Apps natürlich auch für andere infektiöse Krankheiten. Höchstwahrscheinlich ist die aktuelle Pandemie auch ein Anlass, um an verbesserten Technologien zu arbeiten, die beispielsweise eine genauere und zuverlässigere Berechnung der Distanz zwischen zwei Endgeräten zu ermöglichen.
Die COVID-19-Pandemie hat nun aber erstmals auch die Wichtigkeit von Smartphones und Apps für den Gesundheitssektor in der breiten Öffentlichkeit klar gemacht. Smartphones und andere mobile Endgeräte, mit all ihren Sensoren und Kommunikationsschnittstellen, werden in Zukunft sicherlich noch eine wichtigere Rolle bei der Erkennung von Krankheiten, aber auch in deren Bekämpfung einnehmen.
The post Corona Apps & Contact Tracing: Alles, was Sie wissen müssen appeared first on QuickBird Medical.
]]>The post Interoperabilität in der Medizin: An einfachen Beispielen erklärt appeared first on QuickBird Medical.
]]>Das hat allerdings weniger etwas mit Technik zu tun als viel mehr mit menschlicher Koordination. Hersteller medizinischer Geräte stimmen sich bei der Entwicklung oft nicht darüber ab, auf welche Weise diese Geräte Daten austauschen. Das passiert entweder aus mangelnder Kommunikation heraus oder um Kunden an die eigene Marke zu binden.
In der Folge hat jedes Gerät und jede Medical App eine andere Schnittstelle zum Datenaustausch und verwendet einen anderen Standard. Es ist wie beim Turmbau zu Babel: Wenn jeder eine andere Sprache spricht, kann jeder noch so schlaue Sätze sagen – niemand wird etwas davon verstehen.
Wie Sie die Interoperabilitätsanforderungen an eine digitale Gesundheitsanwendung (DiGA) konkret umsetzen, erfahren Sie in diesem Artikel.
Inhaltsverzeichnis
1. Ebenen der Interoperabilität
Durch die zunehmende Digitalisierung und Vernetzung spielt Interoperabilität in der Medizin eine immens wichtige Rolle. Es lohnt sich daher genau zu verstehen, was mit diesem Begriff gemeint ist und welche Aspekte Sie dabei berücksichtigen müssen. Grundsätzlich bezeichnet man mit Interoperabilität
die Fähigkeit verschiedener Systeme miteinander zusammenzuarbeiten.
Man unterscheidet zwischen vier Ebenen:
- Strukturelle Interoperabilität
- Syntaktische Interoperabilität
- Semantische Interoperabilität
- Organisatorische Interoperabilität
Das klingt durch die Fachsprache komplizierter als es ist. Im Folgenden erklären wir anschaulich, was unter diesen Ebenen zu verstehen ist.
1.1 Strukturelle Interoperabilität
Die Grundvoraussetzung, um einen Datenaustausch zwischen medizinischen Geräten (bzw. Medical Apps) überhaupt zu ermöglichen, ist eine Datenverbindung zwischen diesen Geräten. Das kann ein einfaches Kabel sein, welches zwei Geräte miteinander verbindet. Natürlich muss jedes Kabelende auch in die Buchse des jeweiligen Geräts passen. Wenn die Anschlüsse nicht kompatibel sind, nützt das beste Kabel nichts.
Diese technische Voraussetzung, überhaupt Daten von einem Gerät zu einem anderen schicken zu können, wird als strukturelle Interoperabilität bezeichnet. Dazu zählen standardisierte Anschlüsse wie USB, aber auch Datenprotokolle, in denen festgelegt ist, wie Computer miteinander Daten austauschen, z.B. das im Internetprotokoll HTTP.
Ein anschauliches Beispiel aus dem Alltag:
Wenn Sie mit einem Freund kommunizieren wollen, dann müssen Sie natürlich zunächst die technische Voraussetzung dafür schaffen, dass dieser Sie hören kann und umgekehrt. Sie können die strukturelle Interoperabilität herstellen, indem Sie sich z.B. beide in denselben Raum begeben oder indem Sie sich jeweils ein Smartphone besorgen und Ihre Nummern austauschen oder auch eine Chat App verwenden.
1.2 Syntaktische Interoperabilität
Wenn die strukturelle Interoperabilität gewährleistet ist, heißt das noch nicht, dass auch eine Kommunikation stattfinden kann. Es bedeutet lediglich, dass Datenströme, also Pakete aus Nullen und Einsen, zwischen den Geräten ausgetauscht werden können. Diese haben aber an sich noch keine Bedeutung und können je nach Standard unterschiedlich interpretiert werden.
Die Bitfolge 11110000 10011111 10010010 10001010 kann z.B. die für die Zahl 504.623.697 stehen oder für das Emoji 💊. Die Bitfolge 01001000 01100101 01110010 01111010 kann für das Wort Herz stehen oder auch für eine Reihe von Puls-Messwerten: 72, 101, 114, 122.
Durch syntaktische Interoperabilität muss daher bei der Entwicklung sichergestellt werden, dass einzelne Informationseinheiten richtig erkannt werden – oder mit anderen Worten: dass die kommunizierenden Geräte dieselbe Sprache sprechen. Dafür gibt es allgemeine Datenstandards, wie etwa XML und CSV, sowie spezielle medizinische Standards wie HL7.
Beispiel:
Angenommen, Ihr Freund liest Ihnen eine Geschichte vor aus einem Buch. Er spricht Koreanisch und Sie Deutsch. (Sie selbst sprechen kein Koreanisch.) Wenn Sie im selben Raum sind, hören Sie ihren Freund zwar (d.h. die strukturelle Interoperabilität ist hergestellt), Sie können aber nicht einmal heraushören, welche Laute zusammen ein Wort ergeben, weil Sie die Sprache nicht kennen.
Wenn man Sie bitten würde, einfach nur die Worte aufzuschreiben, die Sie hören und zwischen jedem Wort ein Leerzeichen zu lassen, würden Sie sehr wahrscheinlich an dieser Aufgabe scheitern. Syntaktische Interoperabilität würde hier bedeuten, dass Sie in der Lage sind, die Wörter exakt so zu Papier zu bringen, wie sie in dem Buch Ihres Freundes stehen und dass Sie darüber hinaus auch erkennen, zu welcher Wortart jedes dieser Worte zählt (z.B. Substantiv, Verb, Adjektiv usw.).
1.3 Semantische Interoperabilität
Wenn nun die einzelnen Informationseinheiten in einem Datensatz richtig erkannt wurden, muss man im nächsten Schritt der Entwicklung sicherstellen, dass der Empfänger unter den Informationseinheiten auch dasselbe versteht wie der Sender.
Beispiel:
Das Wort ICE bedeutet im Englischen z.B. Eis, im Deutschen dagegen versteht man darunter im Allgemeinen einen Schnellzug.
Genauso müssen medizinische Geräte dasselbe Verständnis von digitalen Informationen haben. Wenn z.B. eine Medical App die Daten 72, 101, 114, 122 von einem Messgerät empfängt, muss klar sein, ob es sich dabei um die Pulsmesswerte eines Tages oder die Körpergewichtswerte eines Jahres handelt. Deshalb wird die semantische Ebene der Interoperabilität benötigt, die ein gemeinsames Verständnis der Informationseinheiten der beteiligten Systeme sicherstellt.
Beispiel:
Haben Sie schon einmal mit einem digitalen Sprachassistenten gesprochen? Alexa, Siri oder Google Home zum Beispiel? Dann kennen Sie bestimmt den Satz, „Entschuldige, aber ich habe dich nicht verstanden.“ In diesen Fällen hat der Sprachassistent zwar oft Ihre Worte verstanden, konnte ihnen aber keinen Sinn zuordnen. Die semantische Interoperabilität zwischen Ihnen und Ihrem Sprachassistenten ist also oft nicht gegeben, da die Technik bislang nur die Semantik einfacher Sätze versteht.
1.4 Organisatorische Interoperabilität
Die letzte Ebene ist die organisatorische Interoperabilität. Hier geht es darum, Prozesse systemübergreifend aufeinander abzustimmen. So muss zum Beispiel sichergestellt werden, dass Ärzte und medizinisches Personal über die entsprechenden Berechtigungen verfügen, um auf Patientendaten zugreifen zu können. Im Kern geht es hier also um definierte Rollen, Datensicherheit und standardisierte Arbeitsabläufe, damit mit den erhobenen Daten auch effizient gearbeitet werden kann.
Beispiel:
Wenn Ihnen Ihr Arzt eine Diagnose oder eine Rechnung per Post schickt, möchten Sie natürlich nicht, dass der Briefträger diese lesen kann. So geht es vielen anderen Menschen auch – nicht nur im medizinischen Bereich. Die Post hat daher einen Prozess etabliert, der sicherstellt, dass nur berechtigte Personen Einsicht in Ihre Post nehmen können: Der Sender steckt die Post in einen Briefumschlag und verklebt diesen. Erst der Empfänger öffnet ihn wieder, wodurch die Datensicherheit gewährleistet wird. Der Briefträger hat in diesem Prozess eine andere Rolle, welche nicht über die Berechtigung verfügt, den Briefinhalt einzusehen.
2. Interoperabilitätsstandards im medizinischen Bereich
Um die größtmögliche Interoperabilität zwischen medizinischen Geräten zu erhalten, ist es natürlich wünschenswert, möglichst wenige Standards zu etablieren. Leider ist dies im medizinischen Bereich nicht der Fall. Es gibt auf den verschiedenen Ebenen viele verschiedene Standards, von denen die meisten gleich mehrere Ebenen der Interoperabilität abdecken, allerdings nicht alle. Dazu zählen zum Beispiel die Standards HL7, SNOMED CT, IEEE 11073 und DICOM. Aufgrund der großen Anzahl an Standards ist es nicht leicht, den richtigen für Ihre medizinischen Geräte oder Anwendungen zu finden. Einen Überblick über diese Standards werden wir Ihnen in unserem nächsten Artikel geben.
3. Fazit
Interoperabilität ist und bleibt ein wichtiger Faktor bei medizinischen Geräten und Medical Apps, ohne die keine brauchbaren Daten zwischen verschiedenen Geräten ausgetauscht werden können.
Speziell in Deutschland ändert sich im Bereich der Gesundheitswesen-Digitalisierung gerade viel durch neue Gesetze wie das DVG, DiGAV oder das Patientendaten-Schutz-Gesetz.
Die Interoperabilität wird aktuell immer mehr in den Vordergrund gerückt. Hersteller sollten diese folglich bei der Entwicklung von Anfang an mitdenken und dabei alle vier Ebenen berücksichtigen. DiGA-Hersteller erfahren hier, wie sie die Anforderungen an Ihre App erfüllen.
The post Interoperabilität in der Medizin: An einfachen Beispielen erklärt appeared first on QuickBird Medical.
]]>The post Leitfaden zur DiGAV: Digitale-Gesundheitsanwendungen-Verordnung appeared first on QuickBird Medical.
]]>Doch welche Anforderungen muss eine DiGA eigentlich erfüllen? Wie läuft die Antragsprozedur ab? Was kostet das Ganze? Derartige Fragen beschäftigen aktuell viele Unternehmen, die eine Medical App bzw. Medical Software planen. Die Digitale-Gesundheitsanwendungen-Verordnung (DiGAV) hat das Ziel derartige Themen zu regeln. Sie ist am 20. April 2020 in Kraft getreten.
Die DiGAV gibt sehr genau Aufschluss, welche Anforderungen an Apps gestellt werden. Wir sind als Software-Dienstleister spezialisiert auf die Entwicklung von Medical Apps. Daher ist das DVG und die DiGAV für viele unserer Kunden ein großes Thema. In diesem Artikel behandeln wir die wichtigsten Inhalte der DiGAV. Falls Sie die Entwicklung einer digitalen Pflegeanwendung (DiPA) planen, lesen Sie unseren DiPA-Leitfaden.
Inhaltsverzeichnis
1. Links zur DiGAV
Vorab finden Sie hier die DiGAV in verschiedenen Formaten, falls Sie bestimmte Dinge dieses Leitfadens später im Detail nachschlagen möchten:
- DiGAV online abrufbar als HTML
- DiGAV Inhaltsverzeichnis online abrufbar als HTML
- DiGAV als PDF
- DiGAV als EPUB
Falls Sie nach diesem Artikel detailliertere Infos zu bestimmten Themen benötigen, lohnt sich ein Blick in den offiziellen DiGA-Leitfaden des BfArM (141 Seiten).
2. Inhalte der Digitale-Gesundheitsanwendungen-Verordnung (DiGAV)
Wichtig: Alle Angaben in diesem Artikel sind ohne Gewähr. Wir sind keine Anwälte, die juristische Beratung anbieten. Inhalte dieses Artikels sind das Ergebnis von intensiven Recherchen, die natürlich dennoch Fehler enthalten könnten.
Im Folgendem werden wir alle Themenblöcke der DiGAV strukturiert durchgehen, um Ihnen einen besseren Überblick zu verschaffen. Jeden Themenblock der DiGAV fassen wir hier zusammen und zeigen auf, wo Sie ggf. mehr Informationen zu dem jeweiligen Paragraphen finden können. Hier sind alle inhaltlichen Punkte der DiGAV für Sie auf einen Blick dargestellt (wir haben die Punkte der DiGAV thematisch gruppiert, um ähnliche Punkte gemeinsam zu behandeln):
- Problem, Ziel, Lösung der Verordnung
- Anforderungen an DiGA
- Anforderungen an Sicherheit, Funktionstauglichkeit, Datenschutz, Datensicherheit und Qualität digitaler Gesundheitsanwendungen
- Anforderungen an den Nachweis positiver Versorgungseffekte
- Antrag
- Antragsverfahren beim Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM)
- Antragsinhalte
- Beratung durch das BfArM
- Inhalte und Veröffentlichung des Verzeichnisses für digitale Gesundheitsanwendungen
- Gebühren und Auslagen
- Anzeige wesentlicher Veränderungen
- Schiedsverfahren
2.1 Problem, Ziel, Lösungsansatz der DiGAV
Das Problem: Aktuell nutzen Versicherte digitale Gesundheitsanwendungen meist komplett auf eigene Kosten. Außerdem haben Patienten keine strukturierten Informationen zu Leistungen und Qualität von digitalen Gesundheitsanwendungen, oder ob die Anwendung gewisse Anforderungen an Datenschutz und Datensicherheit erfüllt.
Die Lösung und das Ziel: Ein zentrales Verzeichnis für digitale Gesundheitsanwendungen wird aufgebaut. In dieses Verzeichnis (sog. DiGA-Verzeichnis) gelangen nur Anwendungen, die Anforderungen an Sicherheit, Qualität, Datenschutz und Datensicherheit erfüllen. Außerdem müssen sie sog. positive Versorgungseffekte nachweisen. Dafür gibt es ein unabhängiges, strukturiertes und verlässliches Prüfverfahren, das die Einhaltung der definierten Anforderungen dauerhaft sicherstellt. Das Verzeichnis ist nutzerfreundlich und transparent für Patienten zugänglich.
Kommentar unsererseits: Einige, wenige Unternehmen hatten auch zuvor schon mühsam erarbeitete Selektivverträge mit Krankenversicherungen, um die Erstattung Ihrer Anwendungen zu ermöglichen. Dies ist jedoch eher die Ausnahme als die Regel. Außerdem müssen derartige Verträge mit jeder einzelnen Krankenversicherung separat verhandelt werden. Das DVG ermöglicht Ihnen als Hersteller Zugang zu allen Krankenversicherungen über einen standardisierten Prozess.
Patienten kämpfen sich bei der Recherche zu Qualität, Datenschutz und Datensicherheit von Gesundheitsanwendungen aktuell durch einen unorganisierten Zertifikatsdschungel. Das DVG soll den einheitlichen „Gütestempel“ für die Erfüllung all dieser Kriterien liefern.
2.2 Anforderungen an DiGA
Für Sie als Hersteller wohl am interessantesten sind die konkreten Anforderungen, die Ihre Medical App bzw. Medical Software erfüllen muss, um ins DiGA-Verzeichnis aufgenommen zu werden. Die DiGAV enthält u.a. einen sehr konkreten Fragebogen in Anlage 1, der diese Anforderungen im Detail abfragt.
Um hier schon mal einen Überblick zu verschaffen, listen wir die Kategorien aller in der Verordnung genannten Anforderungen auf.
2.2.1 Anforderungen an Sicherheit, Funktionstauglichkeit, Datenschutz, Datensicherheit und Qualität digitaler Gesundheitsanwendungen
Zum ersten stellt die DiGAV Anforderungen an Sicherheit, Funktionstauglichkeit, Datenschutz, Datensicherheit und Qualität digitaler Gesundheitsanwendungen:
- Anforderungen an Sicherheit und Funktionstauglichkeit
- Anforderungen an Datenschutz und Datensicherheit
- Anforderungen an Qualität und Interoperabilität
- Anforderungen an Robustheit
- Anforderungen an Verbraucherschutz
- Anforderungen an Nutzerfreundlichkeit
- Anforderungen an die Unterstützung der Leistungserbringer
- Anforderungen an die Qualität der medizinischen Inhalte
- Anforderungen an die Patientensicherheit
Damit Ihre App im Rahmen des DVG erstattungsfähig ist, muss sie ein Medizinprodukt niedriger oder höherer Risikoklasse sein. Daher müssen Sie viele dieser Kriterien ohnehin schon erfüllen (nach MDR oder MDD). Wie Sie entscheiden können, ob Ihre Anwendung ein Medizinprodukt ist, erklären wir in einem gesonderten Artikel. Dabei hilft Ihnen unser Artikel zur Formulierung der Zweckbestimmung und im Anschluss finden Sie die richtige Risikoklasse in unserem entsprechenden MDR-Leitfaden.
Mehr Informationen zur Entwicklung von Medical Apps finden Sie außerdem auch in einem unserem Medical App Development Leitfaden. Der wirklich relevante Teil im Rahmen des DVG wird für Sie der Nachweis positiver Versorgungseffekte sein, den wir im nächsten Abschnitt erklären.
Mehr Details in Abschnitt 2 (§3 – §7) der DiGAV
2.2.2 Anforderungen an den Nachweis positiver Versorgungseffekte
Zum zweiten stellt die DiGAV Anforderungen an den Nachweis positiver Versorgungseffekte. Sie geht zuerst darauf ein, was mit dem Begriff genau gemeint ist. Danach wird die korrekte Darlegung dieser positiven Versorgungseffekt näher spezifiziert. Die DiGAV geht auch auf Studien zum Nachweis positiver Versorgungseffekte ein. Falls Sie derartige Studien zum Antragszeitpunkt nicht vorliegen haben, müssen Sie außerdem ein wissenschaftliches Evaluationskonzept vorlegen, mit dem Sie zu einem späteren Zeitpunkt die Versorgungseffekte auswerten.
Mehr Details in Abschnitt 3 (§8 – §15) der DiGAV
2.3 Antrag
In der DiGAV wird außerdem erläutert, wie der Antrag zur Aufnahme in das DiGA-Verzeichnis abläuft und welche Inhalte dieser haben sollte.
2.3.1 Ablauf des Antrags
Nach Antragsstellung muss das BfArM dem Antragssteller innerhalb von 14 Tagen den vollständigen Eingang der Antragsunterlagen bestätigen oder die Ergänzung fehlender Unterlagen anfordern. Falls Sie zum Zeitpunkt des Antrags einen positiven Versorgungseffekt noch nicht ausreichend nachweisen können (z.B. durch klinische Studien), können Sie eine Erprobungsphase beantragen. Wie lange diese Erprobungsphase dauert, entscheidet das BfArM.
Spätestens zum Ende der Erprobungsphase müssen Sie einen positiven Versorgungseffekt nachweisen. Wenn Sie dies nicht können, müssen Sie dies ausreichend begründen. Falls Sie sinnvoll zeigen können, warum Sie doch noch mehr Zeit zum Nachweis des positiven Versorgungseffekts brauchen, wird die Probephase optional um bis zu 12 Monate verlängert.
Mehr Details in Abschnitt 4 (§16 und §17) der DiGAV
2.3.2 Inhalt des Antrags
Der Antrag beinhaltet u.a. Angaben zu folgenden Punkten:
- Hersteller und identifizierende Merkmale der DiGA
- Medizinische Zweckbestimmung
- Zugehörige benannte Stelle (falls zutreffend)
- Gebrauchsanweisung
- Zielsetzung, Inhalt und Nutzung der DiGA in einer allgemeinverständlichen Form
- Funktionen der DiGA
- An der Entwicklung beteiligte medizinischen Einrichtungen und Organisationen (falls zutreffend)
- Quellen für umgesetzte medizinischen Inhalte und Verfahren, insbesondere Leitlinien, Lehrwerke und Studien
- Vorliegender oder geplanter Nachweis positiver Versorgungseffekte
- Definition der Patientengruppe
- Vorliegender oder geplanter Nachweis positiver Versorgungseffekte für die definierte Patientengruppe
- Studie(n) zum Nachweis positiver Versorgungseffekte
- Diagnostische Instrumente, die für die Studie zur Ermittlung der Testgenauigkeit genutzt wurden (falls zutreffend)
- Zugehörige herstellerunabhängige Institution (falls zutreffend)
- Erfüllung der genannten Anforderungen an DiGA
- In der DiGA vorgesehene Nutzerrollen
- Qualitätsgesicherte Anwendung der digitalen Gesundheitsanwendung inkusive Ausschlusskriterien für die Nutzung
- Ärztliche Leistungen, die für die Anwendung der DiGA benötigt werden
- Mindestdauer der Nutzung der DiGA
- Standorten der Datenverarbeitung
- Kompatibilitätszusagen wie unterstützte Plattformen und Geräte sowie erforderliche Zusatzprodukte
- Genutzte Standards und Profile zur Herstellung semantischer und technischer Interoperabilität
- Deckungssumme der vom Hersteller für Personenschäden abgeschlossenen Haftpflichtversicherung
- Preise der DiGA
Mehr Details in Abschnitt 1 (§2) der DiGAV
2.3.3 Beratung durch das BfArM
Das BfArM berät Sie als Hersteller über den Verfahrensablauf und den mit dem Antrag vorzulegenden Angaben und Nachweisen. Das beinhaltet insbesondere die Beratung zum Nachweis positiver Versorgungseffekte und der korrekten Anzeige wesentlicher Änderungen (nach Aufnahme in das DiGA-Verzeichnis). Das ist natürlich gebührenpflichtig.
Konkret organisiert das BfArM dafür z.B. Beratungstage und Workshops in Deutschland.
Mehr Details in Abschnitt 6 (§23) der DiGAV
2.4 Inhalte und Veröffentlichung des Verzeichnisses für digitale Gesundheitsanwendungen
Das Verzeichnis für digitale Gesundheitsanwendungen wird im Internet veröffentlicht und soll für mehr Transparenz und Übersicht sorgen. Das Verzeichnis wurde bereits veröffentlicht und ist hier einsehbar.
Über jede DiGA werden u.a. Angaben veröffentlicht zu…
- nachgewiesenen oder nachzuweisenden positiven Versorgungseffekten
- vorgelegten Studien einschließlich eines Verweises auf Veröffentlichungsort der Studien
- Sensitivität und Spezifität der diagnostischen Instrumente der DiGA
- Vergütungsbeträgen
- Mehrkosten
- notwendigen ärztlichen Leistungen
Mehr Details in Abschnitt 5 (§20 – §22) der DiGAV
2.5 Gebühren und Auslagen
Die DiGAV geht konkret auf Gebühren und Auslagen ein. Zusammengefasst können Sie für die Entscheidung zur Aufnahme in das DiGA-Verzeichnis, ganz grob mit Kosten zwischen 3.000 Euro und 10.000 Euro rechnen. Außerdem werden Gebühren für Auskünfte und Beratung genannt. Dies erfolgt abhängig vom Aufwand, und kostet mindestens 250 Euro und maximal 5.000 Euro. Für spätere Änderungsanzeigen nach Aufnahme in das Verzeichnis können für Sie Kosten in Höhe von 1.500 Euro bis 4.900 Euro (pro Änderungsanzeige) entstehen. Auch für eine mögliche Ablehnung oder Zurücknahme des Antrags werden Kostenpunkte genannt.
Mehr Details in Abschnitt 7 (§24 – §33) der DiGAV
2.6 Anzeige wesentlicher Veränderungen
Sobald Ihre Medical App bzw. Medical Software es ins DiGA-Verzeichnis geschafft hat, müssen Sie wesentliche Änderungen an der DiGA frühzeitig anzeigen. Eine Änderung an Ihrer Anwendung könnte dazu führen, dass diese nicht mehr die Anforderungen für die Aufnahme in das DiGA-Verzeichnis erfüllt. Dies würde bei der Anzeige jener Änderung sichtbar werden und kann zur Streichung aus dem DiGA-Verzeichnis führen.
Es müssen allerdings nur Änderungen angezeigt werden, die wesentlichen Einfluss auf die Erfüllung der Anforderungen an Sicherheit, Funktionstauglichkeit und Qualität des Medizinproduktes, der Anforderungen an Datenschutz und Datensicherheit oder den Nachweis der positiven Versorgungseffekte ausüben oder zu Änderungen der Patientengruppen, für die die positiven Versorgungseffekte nachgewiesen wurden, führen.
Außerdem müssen Veränderungen angezeigt werden, die im DiGA-Verzeichnis bekannt gemachte Angaben ändern.
Mehr Details in Abschnitt 4 (§18 und §19) der DiGAV
2.7 Schiedsverfahren
Falls es zu einem Schiedsverfahren kommt, regelt die DiGAV hier den Ablauf und Zusammensetzung der Schiedsstelle.
Mehr Details in Abschnitt 8 (§34 – §42) der DiGAV
3. Fazit
Alle Anforderungen der DiGAV zu erfüllen, ist gar nicht so leicht. Besonders weil jede DiGA ein Medizinprodukt sein muss, kommen nicht unerhebliche Kosten und Aufwände auf Medical App- bzw. Medical Software-Hersteller zu. Die Innovation wird dadurch zwar gefördert, aber man sollte es nicht leichtsinnig als „den leichten Weg zum großen Geld“ ansehen. Wenn Sie es trotz aller Hürden als Hersteller in das DiGA-Verzeichnis schaffen, haben Sie natürlich einen starken kompetitiven Vorteil (gerade weil es nicht einfach ist, dort hinein zu gelangen). Was Ende 2019 in Deutschland begann, wird jetzt auch vom Nachbarn Frankreich übernommen: DiGA werden zum Exportschlager – inklusive eines französischen Äquivalents zum Fast-Track-Verfahren. Mehr über die DiGA-Anforderungen in Frankreich erfahren Sie in diesem Blogartikel.
Auch andere Länder, wie Österreich oder Belgien planen die Einführung eines ähnlichen Konzepts.
Uns hat besonders gut gefallen, dass es eine klare Liste von Fragen gibt, die man zur Aufnahme in das DiGA-Verzeichnis beantworten soll. 124 Fragen, viel konkreter hätte man es nicht machen können. Die Fragen wirken sinnvoll und sind eine ideale Checkliste für die Entwicklung qualitativ hochwertiger Medical Apps. Wenn Sie mehr Informationen benötigen, lesen Sie am besten den aktuellen DiGA-Leitfaden des BfArM.
Falls Sie eine Medical App bzw. DiGA planen und Unterstützung bei der Entwicklung benötigen, setzen Sie sich gerne mit uns in Kontakt und wir besprechen das Ganze zusammen unverbindlich. Wir entwickeln Medical Apps für Unternehmen und Forschungseinrichtungen auf Auftragsbasis.
Weitere Fragen? Rufen Sie uns gerne an (+49 (0) 89 54998380) oder schreiben uns eine Email ([email protected]).
The post Leitfaden zur DiGAV: Digitale-Gesundheitsanwendungen-Verordnung appeared first on QuickBird Medical.
]]>The post Digitale-Versorgung-Gesetz (DVG): Spezifikation Digitaler Gesundheitsanwendungen (DiGA) appeared first on QuickBird Medical.
]]>In diesem Artikel erklären wir, was mit „Ärzte dürfen Apps verschreiben“ gemeint ist und speziell, wie Software-Hersteller ihre App oder Software verschreiben lassen können. Das DVG macht die Finanzierung von Apps nicht nur einfacher, sondern soll den Verhandlungsprozess mit den Krankenkassen auch noch erheblich beschleunigen.
Inhaltsverzeichnis
- 1. Apps verschreiben
- 2. Beschleunigte Markteinführung
- 3. Kriterien für die Aufnahme in das Verzeichnis für digitale Gesundheitsanwendungen
- 4. Fazit
1. Apps verschreiben
Grundsätzlich ist nicht von Apps, sondern allgemein von Gesundheitsanwendungen die Rede. Es geht um Medizinprodukte, die durch digitale Technologien die Erkennung, Überwachung, Behandlung, Kompensierung oder Linderung von Krankheiten, Verletzungen oder Behinderungen unterstützen. Dazu zählen zum Beispiel Apps, die Diabetes Patienten helfen, ihren Blutzucker-Spiegel zu dokumentieren oder Apps, die ihre Patienten an das Einnehmen von Medikamenten erinnern.
Patienten können solche Anwendungen künftig durch Verordnung des behandelnden Arztes oder mit Genehmigung der Krankenkasse beziehen. Der App-Hersteller wird dafür entsprechend monetär kompensiert.
Dafür muss der App-Hersteller aber erstmal seine App beim BfArM (Bundesinstitut für Arzneimittel und Medizinprodukte) einreichen und prüfen lassen. Wenn das BfArM zustimmt, wird die App vorläufig ein Jahr lang von der gesetzlichen Krankenversicherung bezahlt. Sobald auch ein positiver Versorgungseffekt nachgewiesen werden konnte, verhandelt der GKV-Spitzenverband gemeinsam mit dem Hersteller über die Höhe der Erstattungsbeträge durch die Krankenkassen. Vor der dauerhaften Aufnahme gelten die vom Hersteller festgelegten Preise.
2. Beschleunigte Markteinführung
Ein weiterer wichtiger Punkt ist die beschleunigte Markteinführung: Das BfArM entscheidet innerhalb von 3 Monaten über den Antrag nach Eingang der vollständigen Antragsunterlagen. Akzeptiert das BfArM die Anwendung/App, wird sie in das DiGA-Verzeichnis (Digitale Gesundheits-Anwendungen) aufgenommen.
Nach der dauerhaften Aufnahme beginnt die Verhandlungsphase über den Erstattungsbetrag für die App mit dem GKV-Spitzenverband. Auch hier gibt es einen klaren Zeitplan: Sollte innerhalb von sechs Monaten nach Verhandlungsbeginn keine Vereinbarung zustande kommen, legt die Schiedsstelle innerhalb von 3 Monaten die Vergütungsbeiträge fest.
Wenn die Gesundheitsanwendung alle Kriterien erfüllt, sind damit alle Wege für eine zügige Markteinführung geebnet. Das erleichtert insbesondere Startups den ohnehin schwierigen Eintritt in den Gesundheitsmarkt.
3. Kriterien für die Aufnahme in das Verzeichnis für digitale Gesundheitsanwendungen
Akzeptiert das BfArM die Anwendung/App, wird sie in das DiGA-Verzeichnis (Digitale Gesundheits-Anwendungen) aufgenommen. Für die Aufnahme muss die Anwendung laut Aussage des BfArM u.a. folgende Kriterien erfüllen. Alle Details zum Verfahren inkl. der genauen Kriterien zur Aufnahme in das DiGA-Verzeichnis werden in einer ergänzenden Rechtsverordnung behandelt. Diese Rechtsverordnung nennt sich DiGAV (Digitale-Gesundheitsanwendungen-Verordnung). Wir haben zur DiGAV hier einen umfangreichen Leitfaden geschrieben. Hier ist schon mal ein grober Überblick:
#1: Die Anwendung muss ein Medizinprodukt von einer niedrigen Risikoklasse (Klasse I oder IIa) sein und erfolgreich ein Konformitätsbewertungsverfahren nach MDR (Medical Device Regulation) bzw. MDD (Medical Device Directive) absolviert haben.
Dieses Kriterium ist sehr klar und die Erfüllung ist über die MDR bzw. MDD konkret geregelt. Wie Sie entscheiden können, ob Ihre Anwendung ein Medizinprodukt ist, erklären wir in einem gesonderten Artikel. Wenn Sie bereits wissen, dass es sich um ein Medizinprodukt handelt, finden Sie dessen Risikoklasse mit unserem MDR-Leitfaden.
#2: Es müssen positive Versorgungseffekte durch die Anwendung nachgewiesen werden.
Die DiGA muss positive Versorgungseffekte nachweisen. Sollte der Hersteller anfangs noch keine positiven Versorgungseffekte nachweisen können, kann er trotzdem beantragen, dass die digitale Gesundheitsanwendung in das DiGA-Verzeichnis zur Erprobung aufgenommen wird. Mit ausreichender Begründung kann dieser Zeitraum um bis zu 12 weitere Monate verlängert werden. Mehr Informationen dazu gibt es in unserem Leitfaden zur DiGAV.
#3: Die Anwendung wird auf Sicherheit, Funktionstauglichkeit und Qualität geprüft
Da die Anwendung ein Medizinprodukt sein muss, wird dieses Kriterium auf natürliche Weise erfüllt sein. Einen besseren Nachweis von Sicherheit, Funktionstauglichkeit und Qualität als das Medizinprodukte-Gesetz/Medizinprodukte-Verordnung gibt es für Gesundheitsanwendungen aktuell nicht.
#4: Die Anwendung muss Datensicherheit und Datenschutz gewährleisten
Man unterscheidet grundsätzlich zwischen Datensicherheit und Datenschutz. Bei Datensicherheit geht es um technische Schutzvorkehrungen zum Schutz von Daten (vor Viren, Manipulationen, Hackern etc.). Datenschutz beschreibt wie personenbezogene Daten weiterverarbeitet werden können, um vor Missbrauch geschützt zu sein (gesetzlich geregelt).
 Vorgaben für Datensicherheit und Datenschutz bei DiGA
Vorgaben für Datensicherheit und Datenschutz bei DiGA
Bei Datensicherheit und Datenschutz macht das Medizinproduktegesetz und die DSGVO bereits Vorgaben. Das BfArM fragt hier zusätzlich anhand eines Fragebogens konkrete Vorgaben ab, wie sich in der DiGAV einsehen lässt. Spätestens ab 2022 wird sogar eine Zertifizierung gemäß der ISO 27000-Reihe gefordert. Ab 2023 müssen alle DiGA (auch die bereits gelisteten) über ein Datensicherheits-Zertifikat des BSI und ein Datenschutz-Zertifikat verfügen. In diesem Artikel erfahren Sie mehr: Datensicherheits- und Datenschutz-Zertifikate für DiGA
Neben den genannten Kriterien gibt es noch einige weitere Bedingungen, die wir hier nicht näher erläutern. Eine vollständige Liste aller Kriterien finden Sie in unserem Artikel zum Thema „Ist Ihre App eine DiGA?“.
Wenn die Applikation alle Kriterien erfüllt und vom BfArM akzeptiert wird, erstatten gesetzliche Krankenkassen ein Jahr lang die Kosten der Anwendung. Der Hersteller muss während dieser Zeit nachweisen, ob seine App die Versorgung der Patienten bessert. Nach Ablauf des Jahres wird dann evaluiert, ob die Applikation weiterhin im DiGA-Verzeichnis bleibt oder nicht. Grundsätzlich berät das BfArM auch Unternehmen bezüglich der Aufnahme in das DiGA-Verzeichnis (kostenpflichtig).
4. Fazit
Das Digitale-Versorgung-Gesetz ist ein wichtiger Schritt, um in Deutschland auch in Zukunft kontinuierliche Innovation im Gesundheitsbereich zu ermöglichen. Die konkreten Bestimmungen sind ein längst überfälliger Schritt, um die Digitalisierung des Gesundheitswesens voranzutreiben.
Die Markteinführung von Gesundheits-Apps wird dadurch nicht nur erleichtert, sondern auch beschleunigt. Das wird es mehr Unternehmen ermöglichen innovative, digitale Produkte auf den Markt zu bringen.
Natürlich ist nicht jede Gesundheits-App sinnvoll. Es ist daher wichtig, dass der Nutzen jeder Gesundheitsanwendung hinterfragt wird. Dass Kassen für Anwendungen zahlen, die keinen Mehrwert für die Behandlung des Patienten bieten, darf nicht die Folge sein.
The post Digitale-Versorgung-Gesetz (DVG): Spezifikation Digitaler Gesundheitsanwendungen (DiGA) appeared first on QuickBird Medical.
]]>The post Leitfaden für die Entwicklung von Medical Apps: Darauf müssen Hersteller achten appeared first on QuickBird Medical.
]]>Es ist aber gar nicht so leicht, eine Medical App auf den Markt zu bringen, die sicher und konform mit Datenschutz- und Medizinproduktrecht innerhalb der MDR (Medical Device Regulation) ist. In diesem Artikel behandeln wir die wichtigsten Herausforderungen, die auf App-Hersteller bei der Entwicklung einer Medical App zukommen.
Inhaltsverzeichnis
- 1. Besonderheiten bei der Entwicklung von Medical Apps
- 2. Qualitätsmanagement im regulatorischen Rahmen
- 2.1 Medizinproduktklasse und Zweckbestimmung definieren
- 2.2 Qualitätsmanagement-System aufbauen: ISO 13485
- 2.3 Einhalten eines dokumentierten Software-Lebenszyklus: IEC 62304
- 2.4 Risiken identifizieren und analysieren: ISO 14971
- 2.5 Gebrauchstauglichkeit sicherstellen: IEC 62366
- 2.6 Benannte Stellen und der Auditing-Prozess
- 3. Datenschutz
- 4. Datensicherheit
- 5. Softwarequalität
- 6. Kommunikation mit medizinischen Geräten & Wearables
- 7. Unterschiedliche Hardware- und Laufzeitumgebungen
- 8. Fazit
1. Besonderheiten bei der Entwicklung von Medical Apps
Grundsätzlich müssen Sie bei der Entwicklung von Medical Apps natürlich erstmal dieselben Dinge beachten wie bei der Entwicklung von Apps in anderen Bereichen: ein intuitives User Interface, eine skalierbare und flexible Softwarearchitektur, etc.
Es gibt aber einige Anforderungen, die speziell bei Medical Apps besonders wichtig sind:
- Qualitätsmanagement im regulatorischen Rahmen
- Datenschutz
- Datensicherheit
- Software-Qualität
- Kommunikation mit medizinischen Geräten & Wearables
- Unterschiedliche Hardware- und Laufzeitumgebungen
In den folgenden Abschnitten werden wir diese Herausforderungen genauer unter die Lupe nehmen.
2. Qualitätsmanagement im regulatorischen Rahmen
2.1 Medizinproduktklasse und Zweckbestimmung definieren
Wir nehmen in diesem Artikel an, dass Sie Ihr Produkt erst mal auf dem europäischen Markt in Verkehr bringen möchten und fokussieren uns nicht auf die regulatorischen Bestimmungen anderer Märkte (z.B. der FDA in den USA).
Medizinprodukt-Hersteller in Deutschland und anderen EU-Staaten müssen sich an die Vorgaben der MDR (Medical Device Regulation) halten.
Wenn Ihre App ein Medizinprodukt im Sinne der MDR ist, müssen Sie ein Qualitätsmanagementsystem aufbauen und pflegen. Ob das so ist, und welche Risikoklasse es in diesem Fall hat, sollten Sie vor Beginn der Entwicklung unbedingt mit einem Regulatory-Experten abklären (kontaktieren Sie uns diesbezüglich gerne).
Als sehr grobe Daumenregel sind Apps, die als Zweck die Prävention, Diagnose oder Therapie von Krankheiten haben, meist ein Medizinprodukt. Wir haben zu dem Thema „Ist Ihre App ein Medizinprodukt?“ auch einen umfangreichen Leitfaden geschrieben, den Sie hier finden können.
Welche Medizinproduktklasse die App dann hat, hängt u.a. vom Patientenrisiko ab. Je höher der mögliche (indirekte) Schaden durch Ihre App ist, desto höher wird die Medizinproduktklasse sein. Für eine erste grobe Einschätzung kann der folgende vereinfachte Entscheidungsbaum hilfreich sein (basiert auf Regel 11 der MDR).
- Wenn Ihre App Informationen liefert, die zu Entscheidungen für diagnostische oder therapeutische Zwecke herangezogen werden: Klasse IIa
- kann Ihre App jedoch potenziell den Tod oder eine irreversible Verschlechterung des Gesundheitszustands einer Person herbeiführen: Klasse III
- kann Ihre App potenziell eine schwerwiegende Verschlechterung des Gesundheitszustands einer Person herbeiführen: Klasse IIb
- Jede andere App: Klasse I
Auch für die Risikoklassifizierung einer App haben wir einen umfassenden Praxisleitfaden verfasst, welchen Sie hier finden.
2.2 Qualitätsmanagement-System aufbauen: ISO 13485
Wenn Sie eine App als Medizinprodukt auf den Markt bringen möchten, kommen Sie um den Aufbau eines nach ISO 13485 zertifizierten Qualitätsmanagement-Systems kaum herum.
Eine komplette Anleitung für den Aufbau eines Qualitätsmanagement-Systems nach ISO 13485 sprengt den Umfang dieses Artikels. Wir haben hier einen weiteren Leitfaden zu ISO 13485 für Softwarehersteller geschrieben. Daher gehen wir hier vor allem auf die wichtigsten Normen ein, die Sie bei der Entwicklung einer Medical App beachten müssen.
2.3 Einhalten eines dokumentierten Software-Lebenszyklus: IEC 62304
Eine Medical App fällt unter die Definition einer „Medizingeräte-Software“. Daher werden Sie nach der Norm IEC 62304 arbeiten (nicht explizit gefordert, aber der absolute Standard), um das Konformitätsverfahren zu bestehen. Die Norm schreibt fünf Prozesse vor, nach denen die Software (weiter-)entwickelt werden muss:
- Software-Entwicklungsprozess
- Software-Wartungsprozess
- Software-Risikomanagementprozess
- Software-Konfigurationsmanagementprozess
- Problemlösungsprozess von Software
Im Rahmen des Software-Entwicklungsprozesses muss dann eine technische Dokumentation angefertigt werden, z.B. zur Software-Architektur und zu Unit-/Integrations- und System-Tests. Außerdem müssen Sie z.B. Ihre SOUPs (Software of Unknown Provenance) umfangreich dokumentieren. Das sind insbesondere externe Software-Bibliotheken, die Sie verwenden. Davon gibt es bei mobilen Apps erfahrungsgemäß besonders viele.
Man hört gelegentlich immer mal wieder Stimmen, die sagen, dass agile Entwicklung im regulatorischen Rahmen nicht möglich ist. Das ist aber so nicht richtig. Mit einem geeigneten Qualitätsmanagement-System und den richtigen Automatisierungstools ist natürlich auch bei der regulierten Entwicklung von medizinischen Apps agile Entwicklung möglich.
Mehr Informationen zur Norm finden Sie in unserem umfangreichen Artikel zur IEC 62304.
2.4 Risiken identifizieren und analysieren: ISO 14971
Sie sollten sehr genau wissen, wo die Risiken Ihrer App verborgen sind. Wenn Ihre App falsche Werte berechnet, abstürzt oder einfach nur falsch verwendet wird, kann potenziell ein Patient zu Schaden kommen.
Die Norm ISO 14971 beschäftigt sich daher mit dem Risikomanagement von Medizinprodukten. Risiken müssen identifiziert und beherrscht (und ggf. durch zusätzliche Vorkehrungen eliminiert) werden.
Grundsätzlich unterscheidet man zwischen Schaden (z.B. eine Schnittwunde), Gefährdung (z.B. ein Messer) und Risiko (die Kombination aus Wahrscheinlichkeit für das Auftreten des Schadens und dem Schweregrad dieses Schadens). Sie müssen nun für jedes Risiko unter Berücksichtigung der Schaden-Wahrscheinlichkeit und des Schaden-Schweregrads entscheiden, ob es akzeptabel oder inakzeptabel ist. Ob ein Risiko akzeptabel ist, hängt vom Produktnutzen ab. Sie müssen abwägen, ob das Risiko in Bezug auf den Produktnutzen akzeptabel ist. Wenn ein Produkt also z.B. keinen Nutzen hat, ist kein Risiko akzeptabel.
Zur Risikoanalyse gibt es verschiedene Verfahren, wie die Preliminary Hazard Analysis (PHA), die Fault Tree Analysis (FTA), und die Fehlermöglichkeits- und -einflussanalyse (FMEA).
2.5 Gebrauchstauglichkeit sicherstellen: IEC 62366
Ihr User Interface sollte nicht nur schön aussehen, sondern auch Fehler bei der Anwendung ausschließen. Nehmen wir beispielsweise eine App zur Steuerung einer Insulinpumpe: Dem Patienten muss in dieser App unmissverständlich kommuniziert werden, welcher Knopf eine Insulin-Injektion auslöst. Eine Insulin-Überdosis kann im schlimmsten Fall sogar zum Tod führen. Ein versehentliches Auslösen ist daher ein nicht vertretbares Risiko.
Genau damit beschäftigt sich die IEC 62366, die Norm zur Gebrauchstauglichkeit von Medizinprodukten. Viele Todesfolgen bei Medizinprodukten sind nämlich gar nicht unbedingt auf Fehlfunktionen zurückzuführen, sondern auf mangelnde Gebrauchstauglichkeit und daher falsche Anwendung. Gebrauchstauglichkeit bzw. Usability Engineering ist auch Teil des Risikomanagements.
2.6 Benannte Stellen und der Auditing-Prozess
Wenn Ihre App unter die Risikoklasse IIa oder höher fällt, müssen Sie eine benannte Stelle hinzuziehen. Benannte Stellen sind privatwirtschaftliche Unternehmen (wie der TÜV), die akkreditiert wurden, um hoheitliche Aufgaben bei der Zulassung und Überwachung von Medizinprodukten zu übernehmen.
Benannte Stellen prüfen z.B. Ihre technische Dokumentation und auditieren und zertifizieren Ihr Qualitätsmanagementsystem. Hier erfahren Sie mehr über benannte Stellen und den Prozess bis zur Medizinprodukt-Zulassung: Zulassung & Zertifizierung von Software-Medizinprodukten (MDR)
3. Datenschutz
Beachten Sie zuallererst, dass Datenschutz und Datensicherheit zwei unterschiedliche Felder behandeln. Bei Datensicherheit geht es um technische Schutzvorkehrungen, um die Sicherheit von Daten zu gewährleisten (vor Viren, Manipulationen, Hackern etc.). Datenschutz beschreibt, wie personenbezogene Daten weiterverarbeitet werden können, um vor Missbrauch geschützt zu sein.

Vorgaben für Datensicherheit und Datenschutz bei DiGA
Mobile Health Apps oder Medical Apps speichern meist sehr sensible Daten, z.B. Ihre persönliche Krankheitsgeschichte. Ihr Nachbar sollte natürlich ohne Ihre explizite Zustimmung nicht wissen, welche Krankheiten Sie haben oder hatten – ein Unternehmen schon gar nicht. Daher müssen sich App-Hersteller stark damit auseinandersetzen, welche Daten sie speichern wollen. Im Zweifelsfall ist weniger mehr. Außerdem muss die App explizit kommunizieren, welche Daten gespeichert werden, wo sie gespeichert werden, und was mit diesen Daten geschieht. Unternehmen, die ihr Produkt auf den europäischen Markt bringen, müssen sich daher mit der DSGVO auseinandersetzen. Datenschutz im Rahmen der DSGVO ist natürlich kein Thema, das nur medizinische Apps betrifft. Jede App muss DSGVO-konform sein.
4. Datensicherheit
Um Datensicherheit zu gewährleisten, brauchen Sie zuallererst ein sehr versiertes technisches Team. Dieses muss umfangreiches Know-how darüber haben, wo Schwachpunkte einer Software liegen könnten.
Sie schaffen es mit einer Medical App mit unzureichender Datensicherheit evtl. auf den Markt, weil auch ein Auditor dies nur bedingt prüfen kann. Die Konsequenzen von nachträglichen Daten-Leaks sind allerdings drastisch (wie Beispiele von Vivy & Co. zeigen). Leichtfertiges Handeln kann einen massiven Reputationsschaden und weitreichende gesetzliche Schwierigkeiten nach sich ziehen.
Die besondere Herausforderung in diesem Bereich: Datensicherheit erlaubt keine 80-Prozent-Lösung. Während Unternehmen gerne beim Design der Software Kosten sparen, kann das bei Datensicherheit nach hinten losgehen.
Wenn Ihre App auch nur eine einzige, leicht nutzbare Sicherheitslücke aufweist, werden Hacker das womöglich herausfinden. Ab diesem Zeitpunkt sind alle anderen Sicherheitsmaßnahmen irrelevant und der Hacker erhält Zugriff auf die Daten.
Wenn in einem verriegelten Haus nur ein einziges Fenster offensteht, kann ein Einbrecher dennoch spielend leicht eindringen.
Für digitale Gesundheitsanwendungen (DiGA) ist eine Zertifizierung der Datensicherheit nach BSI TR-03161 sogar verpflichtend.
5. Softwarequalität
Eine skalierbare Software-Architektur, strukturierter Code und automatisierte Tests (Unit-, Integrations- und System-Tests) sind wichtige Komponenten, um hohe Softwarequalität sicherzustellen. Für Entwickler äußert sich schlechte Softwarequalität in immensem Mehraufwand bei der Software-Wartung und Integration von neuen Funktionen. App-Nutzer spüren mangelnde Softwarequalität meist durch Fehlfunktionen in der App, Abstürze, Performanz-Probleme oder eine schwer zu bedienende Nutzeroberfläche.
Derartige Mängel sind bei Medical Apps besonders fatal: Während ein Absturz der Facebook-App für Nutzer beispielsweise erstmal „nur“ ärgerlich ist, kann der Absturz oder das Fehlverhalten einer Medical App gefährliche, oder sogar lebensbedrohliche Konsequenzen nach sich ziehen. Die Zuverlässigkeit der Software muss daher sichergestellt werden.
Der Einfluss von Softwarequalität wird gerne unterschätzt, auch weil diese als Außenstehender schwer zu evaluieren ist. Das Resultat sieht man oft erst, wenn es schon zu spät ist. Wir empfehlen daher, Ihren versiertesten Software-Entwickler/Techniker bei der Auswahl von neuen Bewerbern und externen Dienstleistern zurate zu ziehen. Er kann durch gezielte technische Fragen meist schnell ein Gefühl für die Kompetenz des Gegenübers erlangen.
6. Kommunikation mit medizinischen Geräten & Wearables
Medical Apps werden oft für die Steuerung und Überwachung von medizinischen Geräten wie Glucose-Messgeräten verwendet. Die Kommunikation erfolgt dafür meist über Bluetooth, Wifi oder USB. Dabei gilt es u.a. auf folgende Dinge zu achten.
Zuallererst müssen Sie das Kommunikationsprotokoll des Geräts genau verstehen. Ein falscher Befehl kann ein medizinisches Gerät zu falschen Aktionen oder sogar zum Absturz bringen. Sie müssen sehr genau wissen, welche Befehle das Gerät in welcher Form erwartet und wie Ihre App die Daten des Geräts interpretieren sollte.
Das ist aufgrund unzureichender Schnittstellen-Dokumentation oft gar nicht so einfach. Es gibt sehr viele verschiedene Standards und viele Geräte benutzen ein nicht-standardisiertes, proprietäres Kommunikationsprotokoll. Dokumentationslücken sollten Sie daher in direkter Kommunikation mit dem Hersteller des Kommunikationsprotokolls klären. Extensives Testen ist auch hier natürlich unerlässlich. Mehr Informationen, worauf bei der sog. Interoperabilität von medizinischen Geräten bzw. Apps ankommt, können Sie auch in diesem Artikel finden. Wenn Sie die Entwicklung einer digitalen Gesundheitsanwendung (DiGA) planen, finden Sie in diesem Artikel konkrete Unterstützung bei der Umsetzung der Interoperabilitätsanforderungen.
Darüber hinaus müssen Sie jederzeit mit Verbindungsabbrüchen rechnen. Eine Bluetooth-Verbindung kann aus vielen Gründen plötzlich abbrechen. Ihre Software muss damit souverän umgehen können und die Verbindung automatisch wiederherstellen.
Zuletzt sollten Sie sicherstellen, dass die Übertragungssicherheit gewährleistet ist, z.B. durch Datenverschlüsselung und Identitätsüberprüfung von Sender und Empfänger. Ein anderes Gerät kann Ihre übertragenen Daten sonst einfach mitlesen oder sogar manipulieren.
7. Unterschiedliche Hardware- und Laufzeitumgebungen
Sogenannte „Embedded Software“ läuft nur auf einem einzigen Gerät. Die integrierte Software eines Glucose-Messgeräts ist hierfür ein gutes Beispiel. Die Rahmenbedingungen sind also statisch und vorab bekannt. Das ist bei Medical Apps aber nicht der Fall. Eine App muss auf einer Vielzahl verschiedener Smartphones funktionieren. Bei jedem Smartphone variieren u.a. folgende Dinge
- Hardware: Bildschirmauflösung, Kamera, Sensoren etc.
- Betriebssystem und Betriebssystem-Version
- Andere Apps, welche die Funktionsweise der eigenen App beeinflussen (wie z.B. Batterie-Manager)
Das ist vor allem bei Android ein großes Problem: Es gibt eine extrem große Menge an verschiedenen Geräten. Es ist daher unmöglich, die App auf allen Geräten zu testen, auf denen sie potenziell laufen wird.

Wie gehen Sie mit solch heterogenen Umgebungen um? Stark zusammengefasst ist Folgendes wichtig:
- Diszipliniertes Testen: Ihre Software-Tester sollten die App auf jeder möglichen Betriebssystem-Version und auf möglichst unterschiedlichen Hardware-Konfigurationen überprüfen (beispielsweise ein kostengünstiges Smartphone mit langsamer CPU und kleinem Bildschirm, und ein Hochklasse-Smartphone mit 4K-Bildschirm und hochmoderner CPU).
- Schulung des Software-Teams: Die Entwickler müssen darauf achten, keine Annahmen zu machen, die auf der Hardware oder der Betriebssystem-Version des Handys basieren.
- Automatisiertes Testen: Manuelle Tests können oft mehrere Wochen Zeit in Anspruch nehmen, wenn sie auf vielen verschiedenen Geräten durchgeführt werden sollen. Mit automatisierten Tests hingegen können Sie Ihre App innerhalb kurzer Zeit z.B. auf 50 verschiedenen Geräten testen.
- Nutzung von Cloud-Angeboten zum Testen der App: Services wie Amazon Device Farm machen es einfach, die App und die zugehörigen Tests auf 50 verschiedenen Geräten auszuführen. Funktionalität, die auf Sensoren oder z.B. auf Bluetooth basiert, kann wiederum nicht auf diese Weise getestet werden.
8. Fazit
Eine Medical App zu entwickeln, birgt viele Herausforderungen. Mit einem interdisziplinären Team aus versierten Software-Entwicklern, Regulatory-Experten, Testern und Medizinern ist dies aber durchaus machbar.
Jede Hürde ist gleichzeitig immer auch ein Vorteil: Wenn Sie die genannten Herausforderungen bezwingen, haben Sie nur noch wenig Konkurrenz. Die meisten Start-ups schaffen es nämlich leider nicht so weit. Das Ganze sollten Sie daher als Chance sehen, sich einen langfristigen Platz auf dem Health-Markt zu sichern.
Auf diesem Weg unterstützen wir Sie gerne. QuickBird Medical hat bereits eine Vielzahl von Health und Medical Apps umgesetzt. Gemeinsam mit unseren Kunden haben wir diese entwickelt und erfolgreich am Markt platziert. Unsere Apps sind nicht nur sicher und verlässlich, sondern bestehen auch im Audit der benannten Stellen. Für eine kostenlose Erstberatung treten Sie gerne mit uns in Kontakt.
The post Leitfaden für die Entwicklung von Medical Apps: Darauf müssen Hersteller achten appeared first on QuickBird Medical.
]]>