In last week’s post, I discussed the difference between the extrinsic and intrinsic structures of a data set. The extrinsic structure, which has to do with how the data points sit in the data space, is encoded by the vector coordinates of the data points. (And remember that these are not spacial coordinates, but abstract coordinates, so the dimension can be arbitrarily high.) The intrinsic structure, on the other hand, has to do with which data points are near each other. The standard way to encode this a very different kind of structure, which is called either a graph or a network, depending on the context. In fact, each of the two terms comes with its own entourage of terminology for all the different parts of the graph/network.
This structure is very simple, consisting of a number of dots that are called vertices in graph terminology and called nodes in network terminology. Between some of these dots are lines that are called edges in graph terminology or links in network terminology, as in the Figure below. (So a graph is made up of vertices connected by edges, while a network is made up of nodes connected by links.) Network terminology is generally used in situations where you want to think of transporting/sending things along the links between nodes, whether those things are physical objects (road networks and rail networks) or information (computer networks and social networks).

Graph terminology is more often used in situations where you want the edges/links to represent other types of relationships between the vertices/nodes. An example that has gotten some attention recently is the “interest graph” in which the vertices are people and topics, and each edge links a person to a topic that they are interested in. You might say that a social network should really be called a graph since we often think of it in terms of the relationships between people rather than the status updates and tweets that get sent between them. In practice, there’s no precise rule for deciding which terms to use, but luckily it isn’t too hard to keep up with both types of terminology.
As I noted last week, there are some data sets (such as social networks) that have an intrinsic structure, but do not have an obvious extrinsic structure. On the other hand, given a data set with an extrinsic structure, in the form of vector coordinates for each data point (This is the type of data that the blog posts before last week all focussed on.) we can always extract an intrinsic structure by building a graph as follows: For each data point, we will put a vertex in the graph. Then for any two points in the original data set that are close together as vectors/points in space, we will connect them by an edge in the graph. (Notice that I’ve switched to graph terms, rather then network terms.)
Now, there are many ways we could do this. First, note that the distances between points are highly dependent on the way that we normalize the data, and we’ll get very different results if we use different normalization schemes. This is a major issue, but there are no general rules for picking the right way to normalize, so I’m going to gloss over this point for now. However, I should note that it is very closely related to the topic of feature selection/engineering, which I’m planning to discuss in future posts.
Once we’ve normalized the data, there are two basic ways to decide what we mean by “close enough”: The first way is to pick a number d and connect any two points whose vector distance is less than d. The second way is to pick a number K and connect each data point to the K other points that are closest to it. Depending on the data set, these two approaches may produce very different results. The second of these methods seems to be more popular in practice because each vertex in the resulting graph has roughly the same number of edges coming out of it (which has some computational benefits.)
We can also augment the basic graph structure by recording numbers called weights that indicate how far apart two data points connected by an edge actually are. The weight between to vertices should be closer to 1 for closer data points and closer to 0 for farther away points. When the points get far enough away that we don’t have an edge at all, we can think of the weight as being equal to zero. These weights are sometimes thought of as the similarity between the two vertices connected by each edge. A common formula for the weight of an edge is the Gaussian function of their distance, which has the property we want: The Gaussian function is equal to 1 when the distance is zero and approaches zero as the distance increases.
There are a number of very different ways in which researchers analyze graphs/networks. One approach, which is more closely associated with the network side of things than the graph side, focuses on understanding individual nodes and how they are related to the entire network. For example, there is the question of centrality: Which node would we expect to have the most traffic flowing past it, whether that means an overloaded server on a computer network, or the most important person in a social network.
However, when thinking of graphs/networks as representing the intrinsic structure of a data set, the focus tends to be on a different type of question. These have to do with thinking of the graph/network as a single geometric object, or as a collection of points sampled from an unseen probability distribution. Latt week, I mentioned the clustering problem, which mostly has to do with the intrinsic structure of a data set. That’s what I’ll focus on in the next few posts. But for this week, I want to demonstrate how we can translate certain algorithms from the setting of vector data sets to graphs.
Modeling algorithms like regression and SVM are difficult to translate to graphs because they rely heavily and explicitly on the extrinsic/vector structure of the data set. However, there are two algorithms we’ve seen that only use the distances between the points: K-nearest neighbors and K-means.
Above, we used vector distances to construct graphs, but now we’ll need to go the opposite way, defining distances between the vertices in a graph. Note that this will include defining distances for far away points, which I’ve been saying is not part of the intrinsic structure. However, this is still different from defining an extrinsic/vector structure because, for example, we still won’t have a notion of angles/dot products.
For an unweighted graph, such as a graph defined by a social network, we will define the distance between two points as being the smallest number of edges that you have to cross to get from one vertex to the other. In other words, this distance is defined by a shortest path (aka a geodesic) between the vertices. For the graph on the left in the Figure above, the distance from the far-left vertex to the far-right vertex will be three. Note that it’s possible that the graph will have two or more pieces (called components) that are not connected by edges. In this case, the distance between vertices in separate components is infinite since there are no paths between them. However, lets not worry about that for now.
If we have a weighted graph then just counting the number of edges between two vertices isn’t exactly what we want to do. Recall that higher weights correspond to closer together vertices, so if two vertices are connected by a long path of high-weight edges, they may be closer together than two vertices connected by a small number of low weight edges. To get around this, we can define the similarity score of a path of edges by multiplying all the weights together. If all the weights are less than one, then this number will go down as we add edges, since adding an edge corresponds to multiplying the total by a number smaller than one. (This method doesn’t work if some of the edges have weight greater than or equal to one.) We can then define the similarity of two vertices to be the smallest similarity score of any path of edges between them.
If we used a Gaussian similarity measure to define the weights on the graph then we can convert the similarity scores back to distances by revering the process, i.e. taking the inverse of the Gaussian function (which involves logarithms and square roots.) However, it turns out that we can translate the KNN and K-means algorithms to the graph setting without explicitly converting the similarities back to distances.
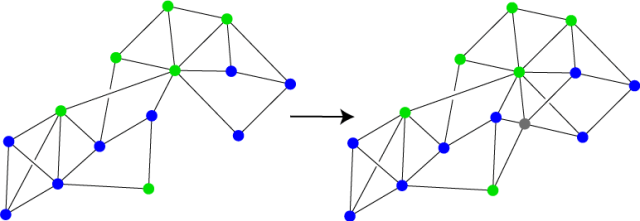
For the KNN algorithm, we will assume that we have a graph in which the vertices are endowed with labels, say blue and green as in the graph on the left in the Figure below. The goal of the algorithm is to choose a label/color for a new data point, which we will insert into the graph in some way, whether by calculating its vector distance to the existing vertices and connecting it to the closest ones, or by using connections that are given to us explicitly, such as the new vertex’s facebook friends. The new vertex is shown in grey in the graph on the right. We can calculate the distance from the new vertex to each of the labeled vertices, choose the K closest ones, and pick the most common label among these to apply to the new vertex. Note that if the graph has weighted edges then we will want to pick the K most “similar” vertices, i.e. the vertices in which the similarity scores to the new vertex are highest.
In the original version of KNN, we noted that this algorithm works by defining a distribution around the data set, closely related to the Voronoi cells that appeared in the Nearest Neighbors algorithm (the precursor to KNN). In the graph, there is no space “around” the data points, so there isn’t an explicit distribution defined by graph KNN. But hopefully, you’re comfortable by now, between looking at kernels and normalization, with the idea that there are many different possible spaces that the data set could be sitting in. When we turn a data set into a graph, we should think of it as still being in one of these, but we don’t know exactly which one. The space is hidden to us, kind of like a black box. We can think of the new points that we add to the graph as being sampled from this hidden space, where the distribution defined by graph KNN lives.
This is a slightly bigger problem with K-means, since the original K-means algorithm relies on calculating “centroids” which are outside the data set. In particular, since we don’t have a vector space that contains the data, we don’t have a notion of taking the average of a set of vertices. (We can’t add vertices together the way we can add vectors.) So for K-means, we will have to replace the centroids with a different notion. There are a number of possibilities, but the simplest is the following: Given a set of vertices in a graph, we will say that the radius of a vertex is the largest distance (or smallest similarity) to any other point in the set. The radius center of a set of vertices in the graph will be the vertex with smallest radius. So we can think of the radius center as being the vertex of the graph/data set that is closest to the hidden centroid in the hidden data space.
With this definition, we can translate K-means directly into the graph setting. We’ll start with K randomly selected vertices, then we’ll repeat the following two steps: First, for each vertex in the graph, associate the vertex to whichever of the selected vertices it is closest to. Second, make a list of the radius centers of each of the resulting sets of vertices. Then we’ll use this new list of radius centers as the selected vertices in the next step. As with vector K-means, this will give us a small number of vertices that are more or less evenly distributed throughout the graph. As with graph KNN, we can’t explicitly see the Voronoi cells produced at each step in graph K-means like we can in vector K-means. However, we can again think of these Voronoi cells as living in some unseen data space that contains the vertices of the graph.
Before I end this post, I want to point out one more thing: If we were to form a graph from a vector data set, using the Gaussian function to define the weights, then convert our similarity scores back to distances as described above, we would find that a lot of the distances had gotten bigger, possibly quite a bit bigger. This is because the distance that comes from the graph, which I’ll call the intrinsic distance, is defined by shortest paths within the graph, i.e. paths that stay “near the data points”. If we could see the underlying probability density function that defined the data set, then these paths would mostly stick to the dark parts of the probability cloud. On the other hand, distance in the original vector space, which I’ll call the extrinsic distance, is defined by shortest paths without restrictions, so these paths can take short cuts that the graph paths cannot.
 If the data set forms a very curved shape, as in the Figure to the left, then these types of distance could be off by quite a bit. In the Figure, the intrinsic distance between the two red vertices, indicated by the green path, is much longer than the extrinsic distance, which is indicated by the dotted blue line.
If the data set forms a very curved shape, as in the Figure to the left, then these types of distance could be off by quite a bit. In the Figure, the intrinsic distance between the two red vertices, indicated by the green path, is much longer than the extrinsic distance, which is indicated by the dotted blue line.
Since these two notions of distance can be so different, this begs the question: Which of them is better for understanding the data set? There is no good answer to this question, partially because both distances depend heavily on the way the data is normalized and a number of other factors. Thus both types of distance could be potentially quite bad. On the other hand, with the correct parameters and normalization, each type of distance will encode very different types of information about the data. In the Figure above, using only the extrinsic structure of the data set would make it hard to distinguish from a Gaussian blob. The intrinsic structure demonstrates the true complexity of the data set by indicating a number of different “fingers”. However, the intrinsic structure doesn’t tell you how these fingers sit with respect to each other in the data space. So ideally, it’s best to consider both the intrinsic and extrinsic structures of a data set by looking at both vector representations and graph representations of the data.







 to the five-dimensional point with coordinates
to the five-dimensional point with coordinates  . If we wanted to give ourselves even more flexibility, we could pick an even higher dimensional kernel, for example by sending the point
. If we wanted to give ourselves even more flexibility, we could pick an even higher dimensional kernel, for example by sending the point  in a nine-dimensional space.
in a nine-dimensional space. . In particular, every monomial in the variables
. In particular, every monomial in the variables  and
and  , such as
, such as  or
or  will appear in one of the entries of this kernel, possibly very far down the sequence.
will appear in one of the entries of this kernel, possibly very far down the sequence.
 . Then we’ll send each data point to the Gaussian function centered at that point. Remember we’re thinking of each of these functions as a vector, so this kernel does what all kernels do: It places each point in our original data space into a higher (in fact, infinite) dimensional vector space.
. Then we’ll send each data point to the Gaussian function centered at that point. Remember we’re thinking of each of these functions as a vector, so this kernel does what all kernels do: It places each point in our original data space into a higher (in fact, infinite) dimensional vector space. and
and  is
is  .
. -dimensional Gaussian kernel, we first choose
-dimensional Gaussian kernel, we first choose  where
where  are the coordinates of the point (in the higher dimensional kernel space) and
are the coordinates of the point (in the higher dimensional kernel space) and  are parameters that define the hyperplane. If we’re using a Gaussian kernel then, thanks to our version of the dot product, the values
are parameters that define the hyperplane. If we’re using a Gaussian kernel then, thanks to our version of the dot product, the values  is either 1 or -1. Then near each data point with label
is either 1 or -1. Then near each data point with label  , the value
, the value  will be very close to 1, while the other values
will be very close to 1, while the other values  will be small, so the sum
will be small, so the sum  will be positive. Similarly, near a point with
will be positive. Similarly, near a point with  , the sum will be negative. Thus if
, the sum will be negative. Thus if  then the decision boundary will separate the positive points from the negative points. In fact, it will carve out a region reminiscent of the Gaussian balls that define the kernel. One example is indicated on the left in the Figure below, where the colors indicate whether the coefficients are positive or negative. As you can see, the result looks something like a smooth version of the nearest neighbors algorithm.
then the decision boundary will separate the positive points from the negative points. In fact, it will carve out a region reminiscent of the Gaussian balls that define the kernel. One example is indicated on the left in the Figure below, where the colors indicate whether the coefficients are positive or negative. As you can see, the result looks something like a smooth version of the nearest neighbors algorithm.



